法规指南
国内法规
GMP 计算机化系统——第四章 验证
第六条 计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。风险评估应当充分考虑计算机化系统的使用范围和用途。
应当在计算机化系统生命周期中保持其验证状态。
第七条 企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系统清单,标明与药品生产质量管理相关的功能。清单应当及时更新。
第八条 企业应当指定专人对通用的商业化计算机软件进行审核,确认其满足用户需求。
在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确保在生命周期内评估系统的质量和性能。
第九条 数据转换格式或迁移时,应当确认数据的数值及含义没有改变
FDA
21 CFR Part 11.10(a): Validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records. (US FDA)
(a) 系统的验证以确保准确、可靠、稳定的预定性能,以及识别无效和改动记录的能力。
21 CFR Part 211.68(a): Automatic, mechanical, or electronic equipment or other types of equipment, including computers, or related systems that will perform a function satisfactorily, may be used in the manufacture, processing, packing, or holding of a drug product. If such equipment is so used, it shall be routinely calibrated, inspected, or checked according to a written program designed to assure proper performance. Written records of those calibration checks and inspections shall be maintained. Fb-p.png
(a) 在药品生产、加工、包装和贮存的过程中,可以使用能够良好执行要求的自动化、机械化或电子设备,包括计算机或其它类型的设备。 为保证此类设备工作性能良好,应根据书面规程对其进行例行校准、检查或核对。应保存校准和检查的记录。 Fb-p.png
21 CFR Part 820 and 61 FR 52602 - Medical Devices; Current Good Manufacturing Practice (CGMP) Final Rule; Quality System Regulation: Software validation is a requirement of the quality system regulation.
General Principles of Software Validation; Final Guidance for Industry and FDA Staff (January 11, 2002): FDA considers software validation to be “confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled.”
FDA Guidance for Industry Blood Establishment Computer System Validation in the User's Facility[8]
FDA Inspection Guides Computerized Systems in
Drug Establishments[9] 内容包括计算机硬件验证&软件验证需要注意的要点。
EU
EMA GMP Annex 11 Computerised Systems——Principle:
The application should be validated; IT infrastructure should be qualified.
该应用程序应进行验证;信息技术基础设施应该是检定的。
Where a computerised system replaces a manual operation, there should be no resultant decrease in product quality, process control or quality assurance. There should be no increase in the overall risk of the process.
计算机系统代替手工操作,应该降低对产品质量,工艺过程控制和质量保证的影响。 不应该有增加在此过程的整体风险。
Excel Spreadsheet 验证
OMCL QM Validation of Computerised Systems Annex 1: Validation of Excel Spreadsheets 2018/03[11] 要求对用于 GMP 用途的 Excel Spreadsheet 的开发、验证和使用过程做验证并进行管控。
根据用途不同将 MS Excel 表格分为两类:
第Ⅰ类:使用电子表格生成一个纸质文件(一般是出自文件中的附件)例如:质量事件的跟踪统计(Tracking Quality metrics)、台账清单(Inventory list)。
第Ⅱ类:需要进行配置或开发以实现更复杂的应用程序功能,包括基本功能和复杂的计算。例如:分析计算(Analytical calculations)、微软关联式数据库使用自定义宏。
ICH
ICH Q7A: Computerized systems used in the manufacture of APIs should be properly developed, validated and maintained to assure data and product integrity. Section 5.4 covers the computer systems validation requirements[13]. (详见 5.4 章节)
WHO
WHO TRS 1019 Annex 3 Good manufacturing practices - guidelines on validation : Appendix 5 Validation of computerized systems 对于计算机化系统验证的介绍与要求。
计算机系统化验证的4个核心
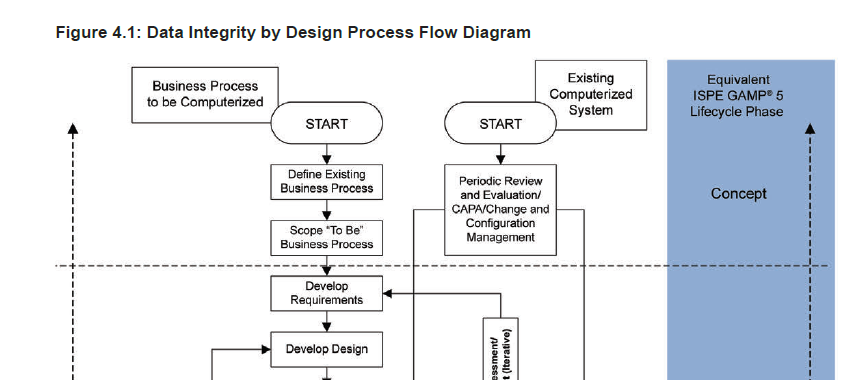
流程: 任何 计算机化系统 都是基于业务流程而设计|配置的,有什么样的流程方面的需求,系统就会设计|配置成什么样子,所以,稍微复杂一些的系统,如果需要起草用户需求,往往是需要来源于业务流程的分析:包括当前Existing Business Process以及所谓的将来的“To-Be” Business Process。
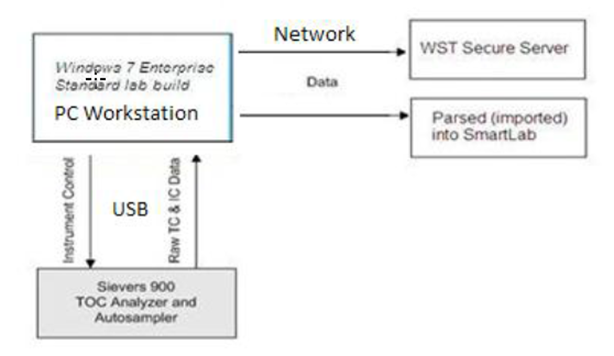
数据: 数据,如果为法规所明确要求,又是以电子形式存在,那就是所谓的电子记录。数据存在的形式是多样的,包括电子形式,纸质和混合的形式。判断数据以哪种形式支持法规活动,也需要结合业务流程去考虑,比如,这台 TOC 分析仪,是以打印出的纸质报告为主数据,还是数据会以电子的形式存储于服务器或者 LIMS 系统,就是不同的业务流程引起的不同的关于主数据形式的判定。数据完整性不仅仅局限于电子形式的数据或法规要求的电子记录,也包括纸质的记录和表单设计
风险: 除了系统的风险之外,针对记录也会有类似的 风险管理 流程,包括识别出哪些场景需要 电子签名 ,系统会产生哪些电子记录,这些电子记录对于 SISPQ 的影响程度,采取哪些控制措施比较合适,相应的控制和管理措施是否有效等等。
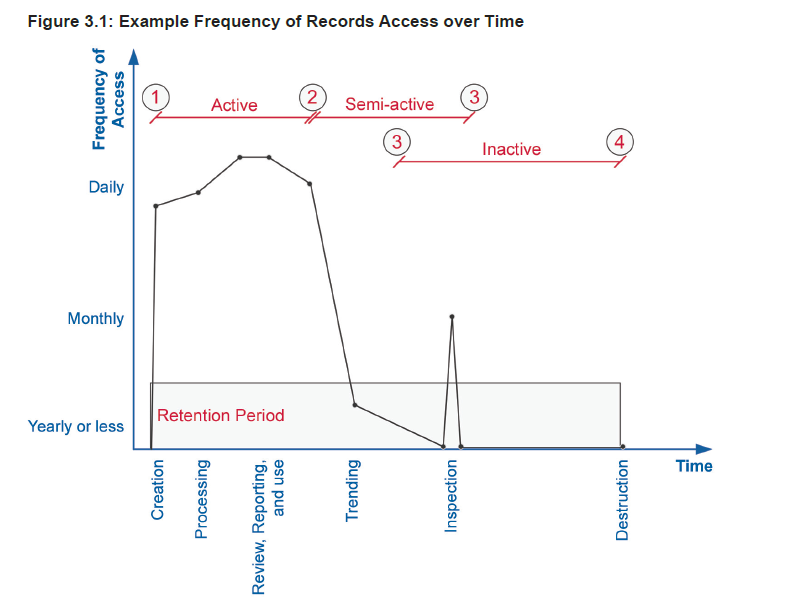
生命周期: 再来一张图,体现的是不同生命周期的数据,访问的频率的高低,数据从不同的维度去理解,有电子数据|纸质数据|混合数据的分法,也有所谓的静态数据和动态数据的分法,或者就是下图的活跃数据|半活跃数据|非活跃数据,不同的阶段,可能界限没那么清晰,也有可能会有一定的Overlap
系统分类
系统软件分类:
类别1——基础设施软件
基础设施中的元件组合在一起,形成一个运行与支持应用程序和服务的完整环境,这个类别中有两种软件:建成的或者市场上可以买到的分层式软件;基础设施软件工具
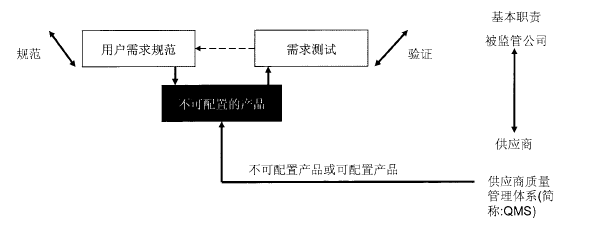
类别3——不可配置软件产品
这个类别包括用于商业目的的可直接使用的现货软件商品。它包括不能通过配置以适应具体业务流程的系统,或者是只可以使用默认配置的可配置系统。这种情形下,配置的系统均可以在用户环境中运行,应基于风险和复杂性来判断使用默认配置的系统归于类别3还是类别4
类别4——可配置软件产品
可配置软件产品提供了标准化的界面和功能,以使配置适合用户的具体业务流程,这通常会需要配置预先设定的软件模块
类别5——定制应用软件
这些系统和子系统的开发是为了满足被监管公司的特殊需要。定制软件的内在风险是非常高的。系统生命周期方法和推广决定都应该考虑到增加的风险,因为用户没有使用经验且缺乏系统可靠性的资料
典型示例和方法
系统硬件分类:
硬件类别1——标准硬件组件
被监管公司所使用的大部分硬件组件都属于本类别。标准硬件组件包括其制造商或者供应商的详细信息以及版本号都应该以文件的形式存档。应验证组件的安装和链接方式是否正确。原装组件的模块、版本号和序列号(如果可以得到)都应该记录下来。
硬件类别2——定制的内置硬件组件
这类硬件是对标准硬件组件的补充。定制的硬件组件应该有设计规范,并通过验收测试。供应商评估应该采用基于风险的方法,并将该评估方法以文件形式存档。在开发定制硬件时,通常都需要进行供应商审计。对使用不同来源的定制硬件组件的组装系统应该进行验证,以确保硬件组件的兼容性。所有硬件配置都应该在设计文件中清晰界定,并验证。
实用的示例
针对软件分类3/4/5类系统如何灵活运用生命周期方法进行说明。
不可配置产品的方法(类别3)
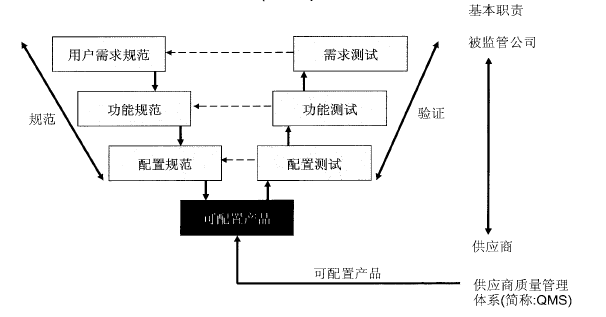
可配置(组态)产品的方法(类别4)
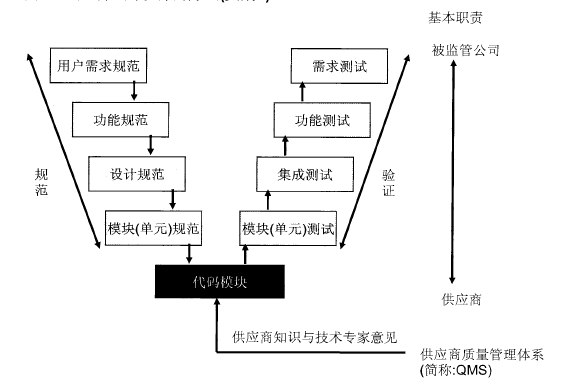
定制应用软件的方法(类别5)
【注】以上内容部分来源于网络,如有侵权,请联系删除