


更多好文,订阅大侠行研专栏( 大侠行研专栏)链接
大侠行研专栏)链接
大侠行研专栏)链接1.(1987 - 1997),“历史的车轮徐徐转动” - 监管开始重视ERES
上世纪九十年代的1987年,随着自动化控制技术(主要参与生产设备管理)和计算机技术(主要参与药物检测及数据分析)引入制药行业。如下图所示,FDA开始逐步认识到了自动化(以PLC逻辑控制器为代表)和信息化(以software软件程序)技术对于药品制造过程和检测过程的重要性,并探索立法引导企业合理使用新技术提升质量,加强管理。最终1997年,FDA发布了 CFR Part 11 电子记录和电子签名,从技术层面定义了一个“新监管”需求和门类,从此“纸质记录 or 电子记录”,“人工操作 or 系统执行”,“线下管理 or 线上流程”等GxP管理上与技术上的实际业务差别开始细分,基于数据风险的风险控制措施开始被人重视!
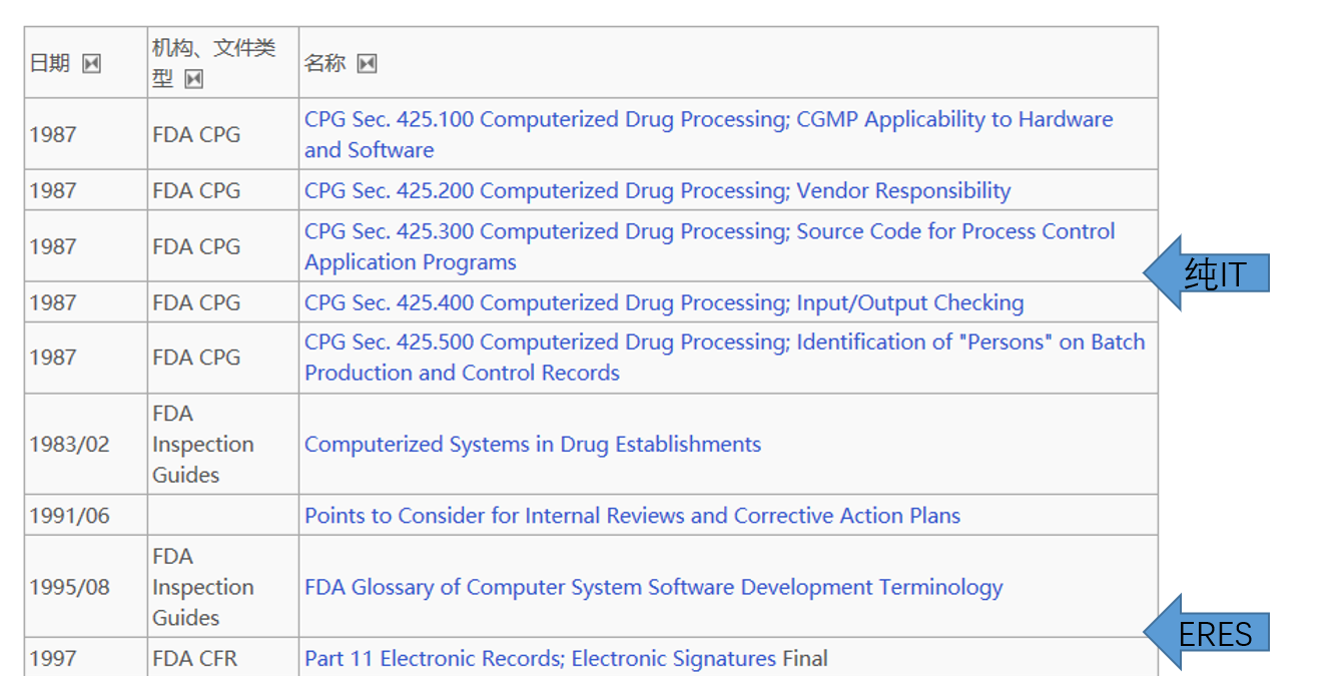
图1. 早期FDA对制药过程(Drug Processing)的自动化&信息化集中在I/O test, 软件硬件检查及批生产检测记录
值得注意的是,同一时间在欧洲,ISPE GAMP于1991年在英国成立,旨在应对美国食品和药物管理局对生产和相关系统GMP符合性的不断发展的期望。GAMP于1994年发布了第一份指导制药工业自动化与信息化意见指南 《Supplier Guide GAMP V1》,成为了日后我们熟知的CSV行业圣经GAMP5的源头!
2.(1997-2017)“达摩克利斯之剑!”-20年让CSV成为行业共识
如果说1997年的FDA Part11 ERES只是让行业知道计算机系统与电子记录对于GxP合规的重要性;那么1997-2017的后20年,制药行业自动化和信息化技术的普及和监管不断发布的新法&指南就让CSV计算机化系统验证 Computerized System Validation这个名词开始烙印在制药行业心中。这20年中,做为制药业我们开始学习IT行业的数据备份(Backup),访问控制(Access Control),灾难恢复计划与业务连续性方案(DRP&BCP);而想进来分一杯羹的仪器设备产商也开始习惯谈Part11 ERES符合性,计算机化系统验证服务以及基于质量的风险管理QRM(Quality Risk Management),一时间“万物皆可CSV”。

图2. 从欧美的计算机化系统法规到中印的相关法规,无论是从时间还是内容,都体现了后发国家向先发国家学习借鉴的这一过程(后面就会有大家喜闻乐见的超车环节了)
从2002年作为先发国家的美国FDA发布了《FDA General Principles of Software Validation; Final Guidance for Industry and FDA Staff FDA对于软件验证的通用指导手册 - 用于制药业与FDA职员》,到欧盟以开创性地以GMP附录形式,将“Computerized System”纳入合规范围,最终到2015年中国正式将计算机化系统纳入中国GMP附录,深陷数据造假2017年印度也发布了自己的计算机化系统系统和电子数据管理指南,由此CSV 计算机化系统验证做为GxP合规要求全部铺开,成为了行业共识!

图3. 2016年1月印度政府在美国首都华盛顿邮报上做的一篇公关广告,形式和内容都耐人寻味。受数据可靠性问题影响,印度制药在美国的信誉下降、商机受损。美国对印度制药质量的担心部分取决于印度社会整体的infrastructure(基础设施、技能人才)水平低、国内医药产业落后。这篇公关广告貌似是招商广告(也许就是,但只有当事者知),但篇头狮子上写的字是Make in India(印度制造),通篇显示都是印度的infrastructure良好,健康产业蒸蒸日上。
3. (2015 - 2022) "旧瓶装新酒?" - 数据可靠性与数据造假的斗智斗勇
《FDA合规官员提醒数据可靠性问题的复苏(FDA合规官员提醒数据可靠性问题的复苏)》-2017年媒体见面会上,美国 FDA 合规办公室高级官员表示,数据可靠性问题不会很快消失,许多品牌药的专利正在到期,巨额资金吸引导致首个提交的压力飙升,这些过去仿制药丑闻滋生的土壤现在又重新显现并且变得更加肥沃。
FDA合规官员提醒数据可靠性问题的复苏)》-2017年媒体见面会上,美国 FDA 合规办公室高级官员表示,数据可靠性问题不会很快消失,许多品牌药的专利正在到期,巨额资金吸引导致首个提交的压力飙升,这些过去仿制药丑闻滋生的土壤现在又重新显现并且变得更加肥沃。数据可靠性风险或者数据造假问题从来不是制药行业新出现的一个问题,而且也不仅仅是GMP制药环节独有的问题,而是GxP药品全生命周期都存在的DI风险,甚至说有时药物研发阶段出现的数据可靠性违规操作的风险更大(毕竟研发管理更为灵活,数据造假后提前上市的收益回报更大)。
而对于计算机化系统,在行业内ERES概念与CSV法规体系逐步成熟后后,随着信息化系统、自动化设备/仪器与电子数据和电子记录的使用日益普及,以“数据完整性杀手”Peter E. Baker连续给多家中印企业发出警告信(数据完整性杀手”Peter E. Baker连续给多家中印企业发出警告信)为爆发点,让原本已经火热的CSV概念,更加占据了药企质量管理的重中之重!
数据完整性杀手”Peter E. Baker连续给多家中印企业发出警告信)为爆发点,让原本已经火热的CSV概念,更加占据了药企质量管理的重中之重!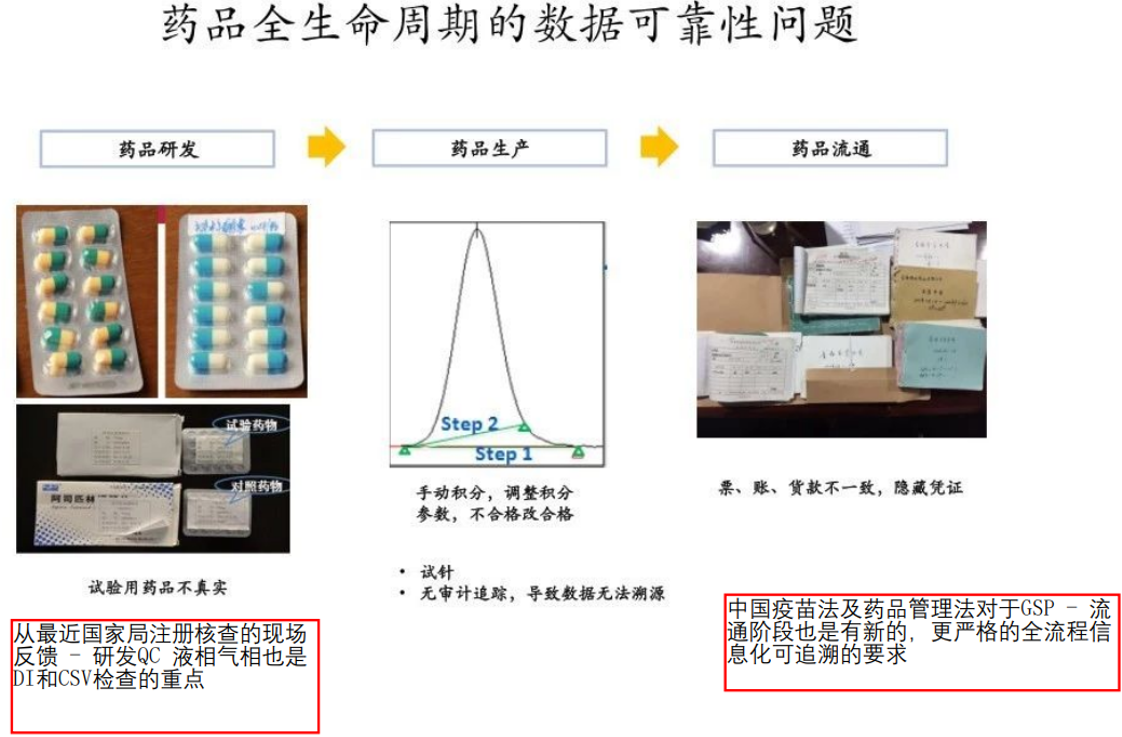
数据可靠性法规指南发展历史
第一个在制药行业具体提出数据可靠性(Data Integrity)要求的指南是2015年1月由英国药品和健康产品管理局(MHRA)发布的《MHRA GMP 行业数据可靠性定义与要求指南》,在这份MHRA的数据可靠性指南当中,数据可靠性被定义为:The extent to which all data are complete, consistent and accurate throughout the data lifecycle. (保证在整个数据生命周期内数据的完整性、一致性和准确性) ,定义里的数据生命周期(Data Life Cycle)指的是从数据产生、贮存、处理、使用、备份、恢复、存档以及数据销毁等各个阶段;同时这份指南也第一次对GMP数据管理提出了后面被广泛引用的数据可靠性(Data Integrity) “ALCOA” 原则(归属至人、清晰可溯、同步记录、原始一致、准确真实)。
2016年5月,世卫组织(WHO) 也正式发布了《良好数据和记录管理实践指南》,其中WHO对数据可靠性再次做了细化和升级- Data Integrity:the degree to which data are complete, consistent, accurate, trustworthy and reliable and that these characteristics of the data are maintained throughout the data life cycle. (数据可靠性是在数据生命周期内完整、一致、准确、值得信赖和可靠以及数据特性被维护的程度) -也就是我们熟知的ALCOA+的数据可靠性原则 - GXP数据和记录不仅要符合ALCOA,而且要符合ALCOA+完整的、一致的、持久的和有效的。
至此业界对于数据可靠性(Data Integrity)的认知基本达成一致,而之后的法规或指南也基本沿用了ALCOA或ALCOA+的概念,并结合自身情况做了一些适当调整:

4. (2022 - ?)下一个CSV&DI热点? 区块链技术防造假,大数据技术优化工艺,AI技术发掘药物靶点
未完待续,等下个假期大侠为你细细道来
