


网页端用户可点击右方目录直接跳转至制定章节
转发本文到朋友圈,添加下方二维码,免费领取【药品注册研制现场和生产现场核查记录表】
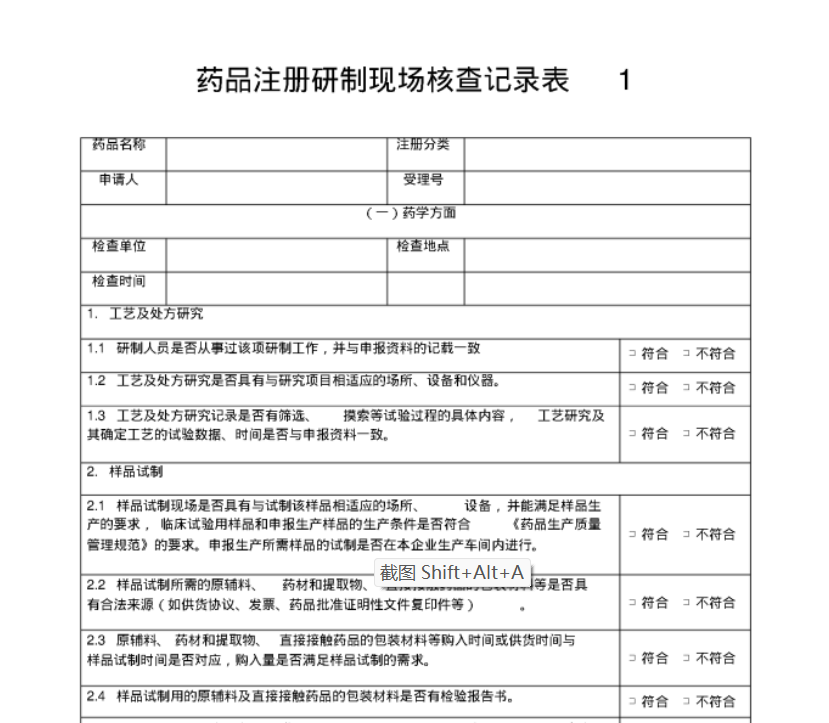

1. 概述
药品注册生产现场核查(生产现场核查),是对药品注册申请的商业规模生产工艺验证、样品生产过程等进行核实,对其是否与申报的或者核定的原辅料及包装材料来源、处方、生产工艺、检验方法和质量标准、稳定性研究等相符合,相关商业规模生产过程的数据可靠性以及是否具备商业化生产条件进行确认的过程。
药品注册生产现场检查(生产现场核查),是指药品监督管理部门对所受理药品注册申请批准上市前的样品批量生产过程等进行实地检查,确认其是否与核定的或申报的生产工艺相符合的过程。
2. 法规指南
2.1 国家局
法律法规和部门规章
第四十五条 药品注册核查,是指为核实申报资料的真实性、一致性以及药品上市商业化生产条件,检查药品研制的合规性、数据可靠性等,对研制现场和生产现场开展的核查活动,以及必要时对药品注册申请所涉及的化学原料药、辅料及直接接触药品的包装材料和容器生产企业、供应商或者其他受托机构开展的延伸检查活动。
药品注册核查启动的原则、程序、时限和要求,由药品审评中心制定公布;药品注册核查实施的原则、程序、时限和要求,由药品核查中心制定公布。
第四十七条 药品审评中心根据申报注册的品种、工艺、设施、既往接受核查情况等因素,基于风险决定是否启动药品注册生产现场核查。
对于创新药、改良型新药以及生物制品等,应当进行药品注册生产现场核查和上市前药品生产质量管理规范检查。
对于仿制药等,根据是否已获得相应生产范围药品生产许可证且已有同剂型品种上市等情况,基于风险进行药品注册生产现场核查、上市前药品生产质量管理规范检查。
第四十八条 药品注册申请受理后,药品审评中心应当在受理后四十日内进行初步审查,需要药品注册生产现场核查的,通知药品核查中心组织核查,提供核查所需的相关材料,同时告知申请人以及申请人或者生产企业所在地省、自治区、直辖市药品监督管理部门。药品核查中心原则上应当在审评时限届满四十日前完成核查工作,并将核查情况、核查结果等相关材料反馈至药品审评中心。
需要上市前药品生产质量管理规范检查的,由药品核查中心协调相关省、自治区、直辖市药品监督管理部门与药品注册生产现场核查同步实施。 上市前药品生产质量管理规范检查的管理要求,按照药品生产监督管理办法的有关规定执行。
申请人应当在规定时限内接受核查。
第四十九条 药品审评中心在审评过程中,发现申报资料真实性存疑或者有明确线索举报等,需要现场检查核实的,应当启动有因检查,必要时进行抽样检验。
第五十条 申请药品上市许可时,申请人和生产企业应当已取得相应的药品生产许可证。
工作文件
CDE 制定公布:药品注册核查启动的原则、程序、时限和要求
- 关于发布《药品注册核查检验启动工作程序(试行)》的通告(2021年第54号),2021-12-20(发布)(自2022年1月1日起施行)
- (
 药品注册核查检验启动工作程序(试行))
药品注册核查检验启动工作程序(试行))
CFDI 制定公布:药品注册核查实施的原则、程序、时限和要求
关于发布《药品注册核查工作程序(试行)》等5个文件的通告(2021年第30号),2021-12-20(发布)(自2022年1月1日起施行)
其他相关
六、现场核查及注册检验。集中受理实施后,国家食品药品监督管理总局新受理的药品注册申请 , 根据药品技术审评中的需求,由国家食品药品监督管理总局食品药品审核查验中心统一组织全国药品注册检查资源实施现场核查,并不再列入2015年7月以来国家食品药品监督管理总局开展的药物临床试验数据自查核查范围。需要进行注册检验的或核查中认为需要抽样检验的,由检查部门按规定抽取样品送中国食品药品检定研究院或省级药品检验机构检验。核查报告和检验报告等,仍按现行规定报送总局药审中心。
第二条 本办法适用于药品监督管理部门对中华人民共和国境内上市药品的生产、经营、使用环节实施的检查、调查、取证、处置等行为。
该办法应不适用于药品注册核查。2018年1月5日原总局发布的《药品检查办法》(征求意见稿)(《药品检查办法》(征求意见稿))适用于食品药品监督管理部门对持有药品批准证明文件的药品上市许可持有人的药品研制、生产环节实施的检查。同时规定国家食品药品监督管理总局负责全国药品检查管理工作,负责药品批准上市前研制过程和药物非临床研究质量管理规范、药物临床试验质量管理规范执行情况的检查,负责药品品种注册和药品上市后变更的检查,负责进口药品的境外检查,负责药品研制和生产有因检查。并且文件正文内容与 国药监药管〔2021〕31号(国药监药管〔2021〕31号)正文差异很大,或不是同一文件。那么,未见后续定稿文件。
《药品检查办法》(征求意见稿))适用于食品药品监督管理部门对持有药品批准证明文件的药品上市许可持有人的药品研制、生产环节实施的检查。同时规定国家食品药品监督管理总局负责全国药品检查管理工作,负责药品批准上市前研制过程和药物非临床研究质量管理规范、药物临床试验质量管理规范执行情况的检查,负责药品品种注册和药品上市后变更的检查,负责进口药品的境外检查,负责药品研制和生产有因检查。并且文件正文内容与 国药监药管〔2021〕31号(国药监药管〔2021〕31号)正文差异很大,或不是同一文件。那么,未见后续定稿文件。境外
药品注册检查检验用申报资料和检查要点
附件 1 :药品研制情况信息表
附件 3 :现场主文件清单
附件 4 :药品注册临床试验研究信息表
附件 5 :临床试验信息表.
说明:适用药品注册核查和药品注册检验的抽样。 新注册办法出台后,暂未有更新的程序和要求。
加强现场检查的情况
2018年5月11日本公告发布之日起 , 对已由省级药品监管部门受理并正在国家药品监督管理局审评审批的化学仿制药注射剂注册申请,国家药品监督管理局将加大有因检查的力度 , 药审中心在严格审评的基础上,根据审评需要提出现场检查需求,由核查中心实施现场检查。
该文件延续“2018年第20号公告”要求。 该文件规定了现场检查品种范围、现场检查工作程序 、相关要求(药品注册申请的撤回、现场检查申请的提交、现场检查申请的撤回、现场检查的要求、纸质材料邮寄地址、联系方式)
指导原则:仿制药一致性评价——现场核查
根据新版《药品注册管理办法》申报资料管理规定,以及《药品生产监督管理办法》和国家药品监督管理局有关职责分工要求,自即日起(2021-12-15),我中心不再接收申请人提交的一致性评价品种现场检查相关整改资料。
十四、受理后,国家食品药品监督管理总局药品审评中心对企业申报资料进行立卷审查。符合要求的,于45日内完成立卷;不符合上述要求的,不予批准,并说明理由。
国家食品药品监督管理总局药品审评中心根据立卷审查情况提出有因检查和抽检的需求,由国家食品药品监督管理总局食品药品审核查验中心统一组织进行对研制现场、生产现场或临床试验数据的有因检查或抽样。 需要检验的,指定有关检验机构。有因检查工作一般在立卷审查结束后60日内完成。
关于请企业在仿制药一致性评价品种受理时提交现场检查有关信息的函
附件2:仿制药质量和疗效一致性评价品种生产情况信息表
附件3:仿制药质量和疗效一致性评价现场主文件清单
仿制药质量和疗效一致性评价生产现场检查指导原则 英文版
仿制药质量和疗效一致性评价有因检查指导原则 英文版
附件2:企业指南:质量和疗效一致性评价仿制药品生产现场检查要求
2.2 省局要求和细则
为落实《药品注册管理办法》(国家市场监督管理总局令第27号)有关药品注册核查的规定,明确药品注册核查实施的原则、程序、时限和要求,核查中心于2020年5月22日起草了《药品注册核查实施原则和程序管理规定(征求意见稿)》。2020年5月22日至6月7日期间征求意见,目前尚未发布定稿规定。 各省局尚未发布更新的药品注册现场核查工作细则。 以下省局要求和细则文件状态不太明朗,仅供参考。
省局研制现场核查要求
2.3 研制和生产现场核查任务信息公告
药品注册申请药学研制和生产现场核查任务信息公告
CFDI 接收来自药品审评中心根据《药品注册管理办法》(市场监管总局令第27号)第48条规定启动的药品注册的药学研制和生产现场检查任务,相关任务信息会予以公告(详见附件)。
根据《药品注册管理办法》,已收到药审中心相关药学研制、生产现场核查告知材料的注册申请人,需尽快登陆 CFDI 网站(www.cfdi.org.cn),通过“药品注册申请人之窗”确认药品注册核查事宜。CFDI 将根据药品注册申请人可以接受现场核查的时间等综合信息,组织安排现场核查工作,其中优先审评品种予以优先安排。
相关药品注册申请人应予以关注,按照相关程序规定,逾期提交者,CFDI 将顺延相关核查任务组织和完成时间,并通知药品审评中心。
以下是信息公告示例:
关注:CDE 关于不再接收一致性评价品种整改资料的通知
根据新版《药品注册管理办法》申报资料管理规定,以及《药品生产监督管理办法》和国家药品监督管理局有关职责分工要求,自即日起(2021-12-15),我中心不再接收申请人提交的一致性评价品种现场检查相关整改资料。
3. 内容推荐
3.1 优秀资源
3.2 优秀文章
3.3 优秀课程
药品注册核查系列课程
 https://www.bopuyun.com/pc/college/333
https://www.bopuyun.com/pc/college/333药品注册现场检查要点及实例分析
https://www.bopuyun.com/pc/college/41药品生产监管要点及缺陷举例
https://www.bopuyun.com/pc/college/367