一、前言
物料与产品管理系统是管理系统的一部分。物料与产品的管理对象、流程管理和基础管理三要素管理是通过放行管理实现的。实施物料与产品放行的主要目的就是保证物料和产品在生产过程中符合相应的药品注册和
2019年12月01日全国人大年月中第十章物料和品放行欧盟GMP》的附录
二、药品“物料与产品放行”相关法律法规文件
1.2019年12月01日全国人大
第二十六条 国家实行疫苗批签发制度
2.2021年月
第一章总
第二章批签发机构确定
第三章批签发申请
第四章
第五章
3. 2011年03月01日NMPA 施行《药品生产质量管理规范》(GMP)
第十章
第二节物料和品放行
3.1.放行对象:
物料:原料、辅料和包装材料等;
产品:中间产品、原料药、原液、待包装批、成品等。
3.2.放行职责:
物料与产品在不同的阶段应有不同岗位职责的人承担不同的职责:物料应当由指定人员签名批准放行所有必需的生产和质量控制均已完成并经相关主管人员签名每批药品均应当由质量受权人签名批准放行疫苗类制品、血液制品、用于血源筛查的体外诊断试剂以及国家食品药品监督管理局规定的其他生物制品放行前还应当取得批签发合格证明
3.3.放行流程:
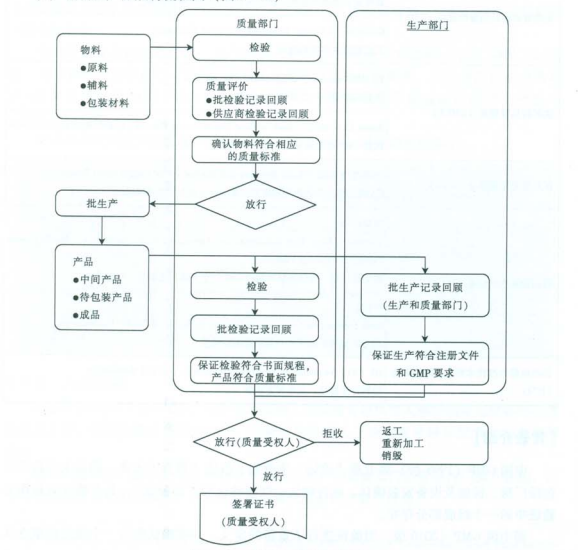
物料与产品放行的主要流程为质量评价和批准放行。
质量评价就是对物料和产品所有相关的原始数据进行评估和批准过程,就是判断物料、产品和过程是否符合质量标准、注册标准和
3.3.1. 物料放行流程
物料的质量评价内容应当至少包括生产商的检验报告、物料包装完整性和密封性的检查情况和检验结果;
3.3.2.产品放行流程
产品放行的质量评价主要包括对批生产记录、批检验记录、环境监测和中间过程等数据(电子数据)进行综合评价。
3.3.2.1. 在批准放行前,应当对每批药品进行质量评价,保证药品及其生产应当符合注册和本规范要求,并确认以下各项内容:3.3.2.3. 已完成所有必需的检查、检验,并综合考虑实际生产条件和生产记录;
4. 欧盟
4.1.范围
4.2.原则
4.3.引言
4.4.通则
4.5.欧洲经济共同体及欧洲经济区生产批产品批的测试和放行
4.6.从第三国进口产品批的测试和放行
4.7.与欧洲经济共同体有互认协议的第三国进口产品的检查和放行
4.8.质量负责人的常规职责
5. FDA《药品生产质量管理规范》(
5.1.严格控制用于药品的标签.
5.2.已发放的一批标签材料,须认真检查其同一性,应与一 批或单批生产记录中说明的标签一致.
5.3. 核对发放的,已使用的及退回的标签,若发现成品数量 与发出的标签数量不符,差额超出根据先前历史水平定下的 数量范围,则需对这些偏差作出评估,按照211∙192要求调 查原因。如果按照211.122(G)(2)执行的100%的贴签正确性 检查,则切割或滚动贴签的平衡可以不做。
5.4. 印有批号或控制号的剩余标签,应全部销毁。
5.5. 退回的标签,如保留应加上证明标志贮存,防止混淆。
5.6. 制订发放标签的详细控制程序,并遵循执行。