


写作前的思考:2022年2月25日王晶同学向我推荐了赤羽雄二的《零秒思考》,这让我意识到自己需要做些什么来精炼自己的知识体系,让自己可以零秒工作。我计划将自己关于微生物相关的经验进行梳理,然后为日后能零秒工作服务。本文主要关于微生物计数法的理解。限于篇幅将会按多部分展开。
一. 微生物技术法的相关法规
1. 了解法规是为了确保自己拥有“入场券”
作为初入制药行业的小白,初始只知道是因为文件规定要做微生物限度检查。久一些知道是为符合药典规定而做。理解更深入一些我们认为是为了保证药品的质量,为了患者的用药安全考虑。
我认为以上想法都是“事后诸葛亮”的杜撰。作为一个制药企业,无论是研发还是生产都是为了盈利。为了有资格进行药品的生产,我们才按法规要求来生产(求轻踩,我不是一个有情怀的人)。制药人中真的能将患者装在心里的人并不多,毕竟我们只是一个打工人,工作是为了更好的生活。
药品是特殊的商品,关乎民生。为了民生和社会稳定,作为监管机构需确保药品可及、安全和有效,从而提出一些对药品质量进行控制的要求。监管机构要求制药企业进行微生物限度检查原因如下:①微生物本身会影响药品的质量:微生物生长繁殖可能或引起药品的腐败变质;②某些微生物本身或其代谢产物会对人体健康造成不良影响,如致病菌或内毒素等。
作为一个制药人在考虑是否进行微生物限度检查或者如何制定标准时,我们可以在考虑钱袋子的基础上以监管者的角度来看问题,这样我们就能更好的做决策。我们只需明白法规要求,保证自己可以拿到入场券,然后在此基础上考虑成本和收益,进行评估后就可以做决定了。
2. 微生物计数法的相关法法规
2.1. 关于微生物计数的相关法规思维导图
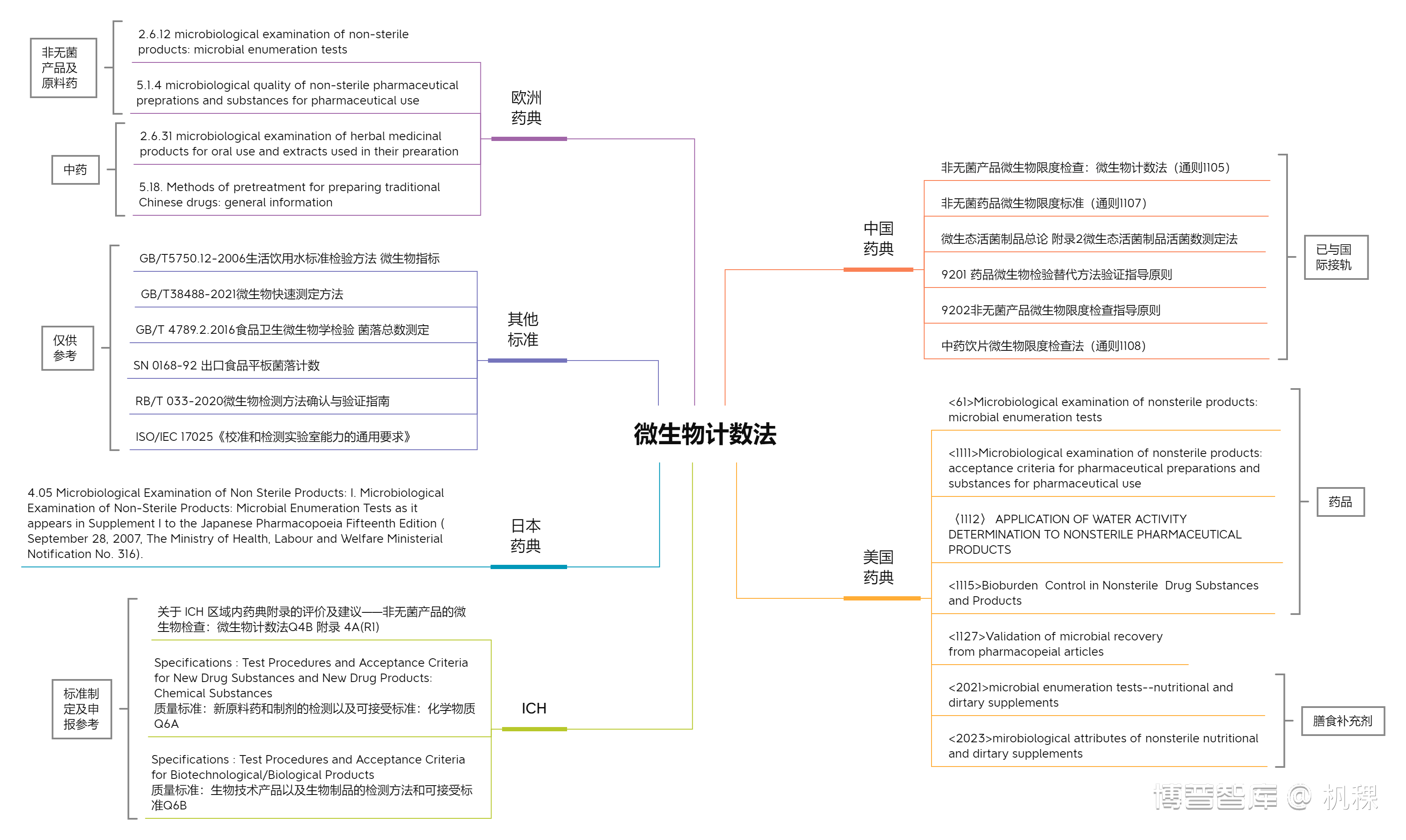
2.2. 关于微生物计数方法法规的使用说明
A. 关于药典的选择
各国药典肯定是我们学习微生物计数法的首要参考。,因为ICH Q4B的存在,对于欧美及日本药典关于微生物计数法的要求区别并不大,建议除关注中国药典外可以适当参考美国药典。其中<1112>、<1115>对于水活度检测和微生物负载控制的描述是特别经典和值得参考的。
需要注意各国药典都将中药制剂进行了更详细的规定。
B. 关于标准制定的参考法规
在研发申报时我们需要确认自己产品是否需要进行微生物限度检查时参考ICH Q6A和ICH Q6B。在确定需要进行微生物限度检查后我们需要考虑看中国药典四部的1107来确定标准制定的基本原则。涉及国外申报的可以再看美国药典的<1111>、欧洲药典的5.1.4、日本药典的G4,其中化药相关制剂的微生物计数方法标准差异不大,但是对于中药制剂相关的标准,各国标准存在些许差异。
除需要参考通则项下规定制定标准外,需特别注意:如各论下有具体规定需要按各论执行。
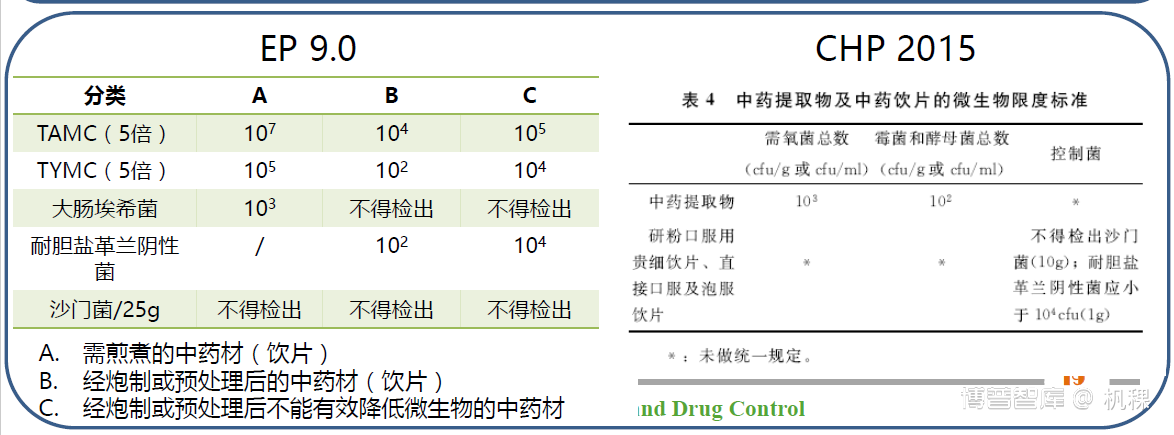
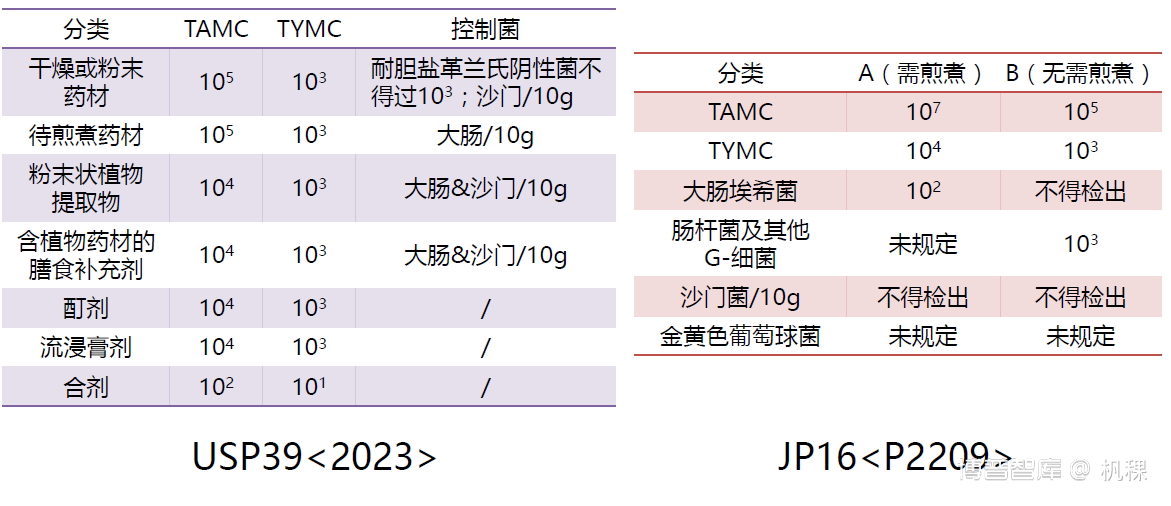
C. 关于其他法规
将部分食品微生物计数法相关和CNAS实验管理的法规列入思维导图,旨在是提醒微生物检验人员,在遇到检验问题或管理问题时我们可以参考他们来拓宽自己的思路,用于借鉴。
2.3. 如何制定一个微生物限度检查标准
2.3.1. 制定微生物限度控制标准的思路:收集基本信息:产品的剂型、给药途径、用药人群、药品特性、产品的竞品信息(特别是仿制药)等;查看药典相关规定(包括通则和各论),确认标准的初步范围;查看相关进口注册标准进行标准的进一步确认,通常标准会收严,特别是控制菌检查项目;结合生产工艺和产品特性确认暂定标准是否满足药品安全、有效和质量稳定的要求;结合现有检测技术确认标准是否可以被正确执行,此处特别强调:标准不仅是数值,其单位同样具有一定的意义。例如1cfu/mL与100cfu/100mL代表的意义并不同,前者检验量可以是10mL也可以是100mL,而后者不能小于100mL。
对于原辅料的微生物限度标准制定也可以参考以上思路进行制定,同时可以结合供应商的建议进行制定。
2.3.2. 我对《中国药典》2020版1107非无菌药品微生物限度标准的理解
2.3.2.1. 制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料应符合无菌检查法规定。
解读:此处需补充一点:对于原辅料需要根据我们的生产工艺和用途来进行确认,而不是照抄药典标准规定;例如氯化钠在无除菌方式的无菌生产工艺中就需要进行无菌检查或者供应商提供给我们的无菌聚山梨酯80不一定需要进行无菌检查。
2.3.2.2. 用于手术、严重烧伤、严重创伤的局部给药制剂应符合无菌检查法规定。
解读:基于用药安全考虑。此处需要注意因为用药环境可能不是无菌的,因此药品使用说明可以根据实际在必要时规定药品需要一次性完成使用。因为我们无法保证使用后依旧是无菌的。
2.3.2.3. 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表1。
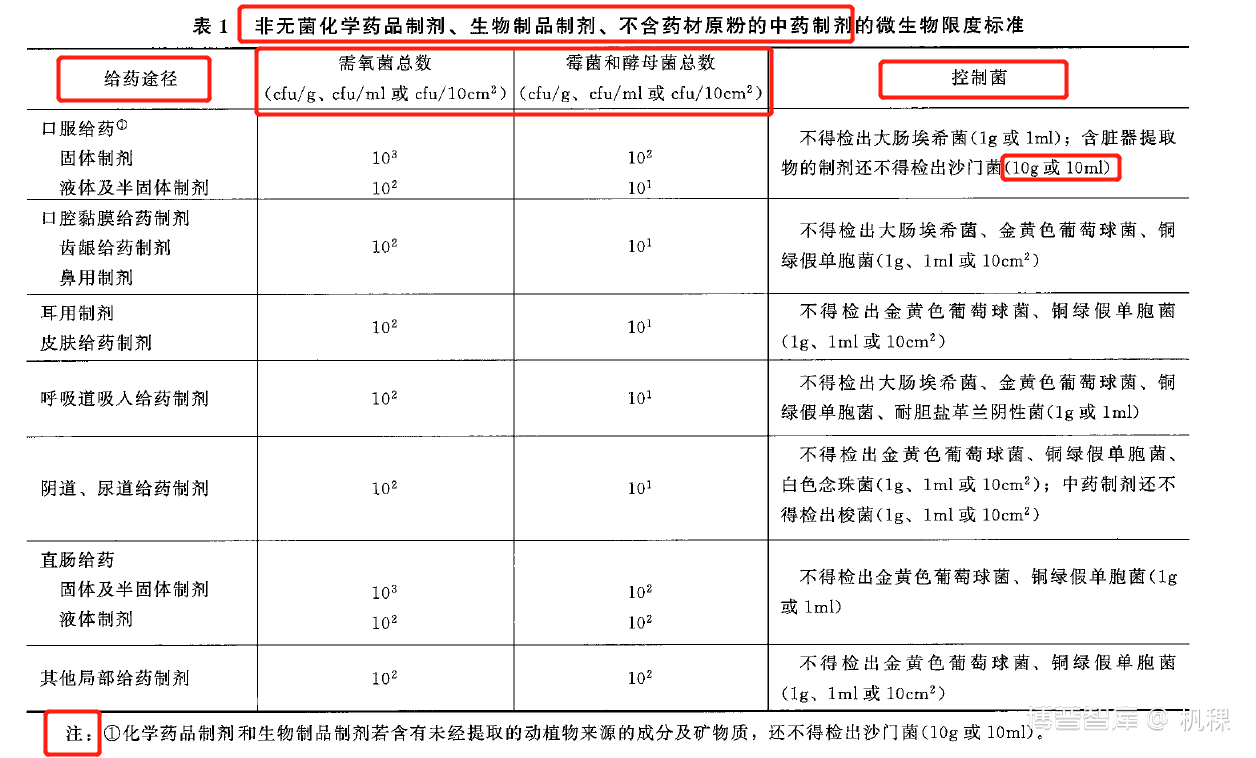
(截图来源于中国药典通则1107非无菌药品微生物限度标准,仅供参考)
解读:对于表1我们需要注意其适用范围是制剂,其次需要注意标红框的内容:
A. 给药途径:给药途径项下包含给药方式和药品物理状态部分内容。口服给药因为经过肠道风险较小而控制最为宽松;对于液体和半固体制剂因为其水活度高更利于微生物生长而控制严于固体制剂。其中在我们进行微生物限度检查标准评估时水活度、渗透压等物理因素也可以是支持我们观点中药论据。
B. cfu/g、cfu/ml、cfu/cm3三个种单位对应三种取样方式:称量(通常为固体)、量取(通常是液体)、取适当面积的待检样品(通常是膜剂、包材等);
C. 控制菌:进行该项检验标准制定时需要特别注意检验量的问题。其次对于大检验量的问题,可以考虑采取薄膜过滤法来解决(需要是液体或者可溶解)或通过等份分瓶的方式来解决(此时会有人问方法学验证的时候是否需要按同样方式进行验证呢?个人认为如可以确保被等份,每份样品都一致的情况下可以只选取其中一瓶加试验菌作为试验组)。
D. 注:在看表格时需要特别注意注。此处备注的意思是动植物来源的成分和矿物质,因为其存在污染沙门菌的风险所以制剂需要增加沙门菌的检验。此处告诉我们在选取原辅料时还需要关注其来源。鉴于此个人建议优先选择化学合成的原辅料,然后选择植物性来源的原辅料,没有选择时再选择动物源原辅料。理由:考虑日常生产稳定性、安全性和降低日常质量监控的成本。
2.3.2.4. 非无菌含药材原粉的中药制剂的微生物限度标准见表2 。

(截图来源于中国药典通则1107非无菌药品微生物限度标准,仅供参考)
解读:理解时参考表1进行理解。需要注意的不同点就是增加了耐胆盐革兰氏阴性菌的检查,在检查时注意定性方法和定量方法的不同。
2.3.2.5. 非无菌药用原料及辅料的微生物限度标准见表3

(截图来源于中国药典通则1107非无菌药品微生物限度标准,仅供参考)
解读:此处控制菌未做统一规定的意思并不是不做要求,需要我们根据原辅料本身的来源、性质、预定用途和生产工艺进行评估。来源,通常表1备注,对于动植物来源或矿物质需要特别关照,如我们不在原辅料阶段进行控制就可能导致制剂的不合格;性质,对于强酸或强碱或杀菌剂等,因为其本身的特点不利于微生物生长甚至杀菌,我们不对其进行微生物限度检查;预定用途,对于我们用于研发小试确定不会用于临床的物料可以不进行微生物限度检查;生产工艺,对于生物制剂上游生产用于配制各种溶液的物料,我们通常会对溶液进行除菌过滤后使用,所以个人认为可以适当放宽控制标准。然而对于一些最终灭菌的注射剂生产用的原辅料我们需要考虑进行大肠埃希菌的检查,而且在原辅料超标出现大量革兰氏阴性菌时我们需要谨慎对待。
2.3.2.6. 中药提取物及中药饮片的微生物限度标准见表4

(截图来源于中国药典通则1107非无菌药品微生物限度标准,仅供参考)
解读:需要注意耐热菌的检查。此处应特指用于中药临床用的中药提取物和中药饮片。对于制剂生产的中药提取物或中药饮片其标准的制定,需要结合生产工艺、存储条件及存储期限等因素进行评估分析,因为生产过程中的高热等情况可以实现微生物的杀灭同时可能产生微生物死亡产物(如内毒素等)。
2.3.2.7. 有兼用途径的制剂
应符合各给药途径的标准。
解读:从严,如从严后控制成本过高可以考虑在说明书中明确使用途径。
2.3.2.8. 除中药饮片外,非无菌药品的需氧菌总数、霉菌和酵母菌总数照“非无菌产品微生物限度检查:微生物计数法(通则1105)”检查;非无菌药品的控制菌照“非无菌产品微生物限度检查:控制菌检查法(通则1106)”检查。各品种项下规定的需氧菌总数、霉菌和酵母菌总数标准解释如下:
101 cfu : 可接受的最大菌数为20;
102cfu : 可接受的最大菌数为200;
103cfU : 可接受的最大菌数为2 0 0 0 ; 依此类推。
中药饮片的需氧菌总数、霉菌和酵母菌总数及控制菌检查照“中药饮片微生物限度检查法”(通则1108)检查;各品种项下规定的需氧菌总数、霉菌和酵母菌总数标准解释如下:
101cfu: 可接受的最大菌数为50;
102cfu: 可接受的最大菌数为500;
103cfu: 可接受的最大菌数为5000;
104cfu: 可接受的最大菌数为50000 ; 依此类推。
解读:因此各种相关培训都已经说过很多次就不再啰嗦。只强调两点:对于内控是如我们规定1000cfu/g时不可解读为2000cfu/g;报告结果时要按照10的n次幂来报告结果。
2.3.2.9. 本限度标准所列的控制菌对于控制某些药品的微生物质量可能并不全面,因此,对于原料、辅料及某些特定的制剂,根据原辅料及其制剂的特性和用途、制剂的生产工艺等因素,可能还需检查其他具有潜在危害的微生物。
解读:此处强调一点,对于制剂是这样的,对于原辅料同样是如此的。示例可以参考2.3.2.5。其中洋葱菌群是需要特别关注的。具体内容见传送门:洋葱伯克霍尔德菌群的那些事(二) - 博普智库 (bopuyun.com)洋葱伯克霍尔德菌群的那些事(一) - 博普智库 (bopuyun.com)
2.3.2.10. 除了本限度标准所列的控制菌外,药品中若检出其他可能具有潜在危害性的微生物,应从以下方面进行评估。药品的给药途径:给药途径不同,其危害不同;药品的特性:药品是否促进微生物生长,或者药品是否有足够的抑制微生物生长能力;药品的使用方法;用药人群:用药人群不同,如新生儿、婴幼儿及体弱者,风险可能不同;患者使用免疫抑制剂和甾体类固醇激素等药品的情况;存在疾病、伤残和器官损伤;等等。
解读:该部分个人以为是建立在历史数据的基础上进行的,核心依旧是保证患者的用药安全和保证产品的质量稳定。
A. 药品的给药途径:关于不同给药途径的危害不同这点,除了直肠给药和局部给药不需要检验大肠埃希菌以外,其他的都需要;需要注意接触粘膜和伤口的都需要控制金黄色葡萄球菌和铜绿假单胞菌。记住以上两点基本可以确认大部分药物的控制菌;
B. 药品的特性,其中前半句很容易理解,后半句对于含抑菌成分的产品有利,但是后半句需要注意不能过度解读,如认为本品抑菌所以不进行控制菌检查是不适宜的,建议只针对潜在不可接受微生物是否需要控制时基于此进行评估;
C. 药品的使用方法这点,需要特别注意多次使用的制剂和具有多种使用途径的药物,从严控制或者在药品开发时就要考虑是否优化处方或通过包装的改善来解决问题(如瓶装换成铝箔等);
D. 用药人群跟各项后面都是针对免疫力低下人群的,需要特别注意有些成人用药改为儿童用药时不仅是剂量的变化及处方的优化,还需要注意是否收严控制微生物标准等。
E. 以上评估方式不仅可以用于我们评估控制菌检查。我们还可以结合工艺、生产环境、运输条件等评估是否进行某些产品的微生物限度检查或是否对不同厂家的同品名的物料是否进行方法适用性试验等。
2.3.2.11. 当进行上述相关因素的风险评估时,评估人员应经过微生物学和微生物数据分析等方面的专业知识培训。评估原辅料微生物质量时,应考虑相应制剂的生产工艺、现有的检测技术及原辅料符合该标准的必要性。
解读:制剂生产工艺我们除注意是否存在吸附、除菌过滤、高温、高渗、干燥等条件外,同样也需要注意促生长的各种情况。
二. 第二部分见下面链接
微生物计数法学习小结(二):方法适用性及日常检验 - 博普智库 (bopuyun.com)
