本周法规主要内容包括:5个指导原则征求意见稿、5个其他征求意见文件、中药饮片特别征集、疫苗特别管理规定、一周其他法规。
一、5个指导原则征求意见稿
本周药品审评中心共发布5个指导原则(征求意见稿):《抗体偶联药物非临床研究技术指导原则(征求意见稿)》、《组织患者参与药物研发的一般考虑指导原则(征求意见稿)》、《注射用两性霉素B脂质体生物等效性研究技术指导原则(征求意见稿)》、《药物真实世界研究设计与方案框架指导原则(征求意见稿)》、《化学仿制药口服调释制剂乙醇剂量倾泻试验药学研究技术指导原则(征求意见稿)》
指导原则征求意见主要内容:
1、《抗体偶联药物非临床研究技术指导原则(征求意见稿)》
2、《组织患者参与药物研发的一般考虑指导原则(征求意见稿)》
3、用两性霉素B脂质体生物等效性研究技术指导原则(征求意见稿)》
4、《药物真实世界研究设计与方案框架指导原则(征求意见稿)》
5、《化学仿制药口服调释制剂乙醇剂量倾泻试验药学研究技术指导原则(征求意见稿)》
二、5个其他征求意见文件
1、2022年7月5日药品审评中心发布了第六十批参比制剂(征求意见)
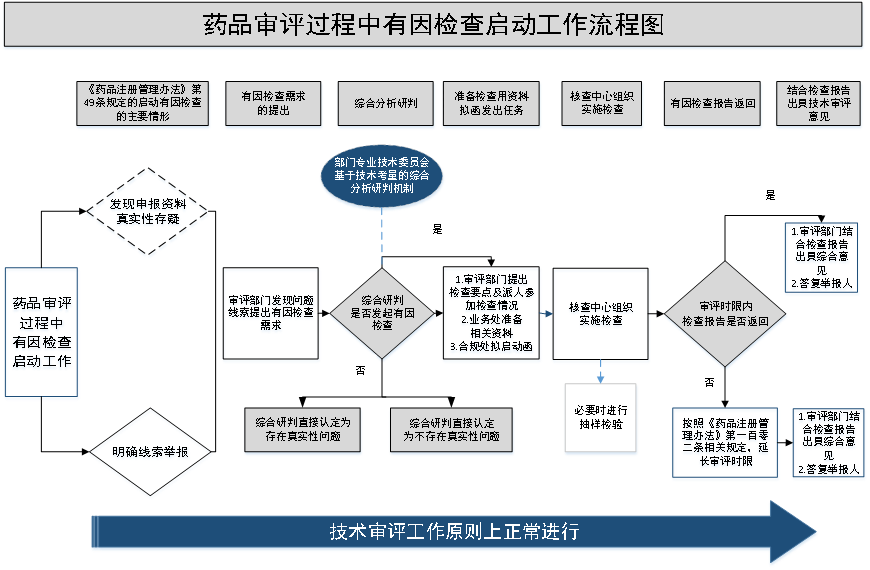
2、《药品审评过程中有因检查启动工作程序(征求意见稿)》
3、《人类辅助生殖技术用医疗器械 器具类产品通用要求(征求意见稿)》
4、药典委对重点研究的品种进行征求意见检测细菌内毒素
5、中检院征求企业参与标准研制工作
三、中药饮片特别征集
2022年7月8日,国家局发布中药饮片特别征集活动,其主要内容包括:中药饮片包装标签管理规定(征求意见稿)、《中药饮片包装标签管理规定(征求意见稿)》起草说明、中药饮片标签内容撰写指导原则(征求意见稿)、中药饮片保质期研究确定技术指导原则(征求意见稿)、《中药饮片保质期研究确定技术指导原则(征求意见稿)》起草说明
四、疫苗特别管理规定
为贯彻落实《中华人民共和国药品管理法》和《中华人民共和国疫苗管理法》等法律法规要求,构建科学、有效的疫苗生产流通监督管理体系,根据疫苗产品特性和疫苗监管要求,依法对疫苗的生产、流通管理活动进行规范,2022年7月8日国家药监局发布了《疫苗生产流通管理规定》,自发布之日起施行。
五、一周其他法规
2022年7月4日:
2022年7月5日
2022年7月6日
更多政策法规请关注博普智库政策法规库哦!点击进入→政策法规库