


写在前面的话:该部分主要结合郑老师讲的资料审核重点发表个人见解,该部分仅为个人观点,欢迎批评指正。

图片来源于网络仅供学习交流用
1.资料审核
开展实验室工作前先进行资料审核,资料审核通过后方可进行实验室工作。关于资料审核郑老师未进一步展开,主要强调了以下五点,蓝字部分是个人思考。
1.1.每一注册复核事项仅接受一次与提交CDE一致的资料审核,需要避免资料缺失或欠完善无法开展实验室工作。
思考:
对于热衷于赶进度的人需特别注意 “每一注册复核事项仅接受一次与提交CDE一致”,不要因为微生物检验方法开发时间长而想着先申报再补充完善。我曾经遇到过某产品的申报因一个微生物检验项目发补了四次,起因就是他们想逐步完善再就是被某第三方坑苦了。
关于资料缺失或欠完善,个人将其原因分为三种情况:
1)因未进行研究导致资料缺失或欠完善,这种情况很少但是不可避免的会出现,这种情况是硬伤,只能回炉重造。
需要特别注意不要有打擦边球的想法,是不是可以质量标准中不体现就不进行研究的想法是不可行的。微生物检查作为安全性检查项目,其质量研究过程必然会涉及,对于参数放行等是强调过程控制并不是不控制,甚至微生物检验相关研究更多。
2)因认识不足导致资料缺失或欠完善,很多项目有时候考虑成本抑或忽视微生物基础知识导致研究不充分进而导致资料确实或欠完善。
某些原料药价格比较昂贵,很多企业就想着法减少检验量或检验项目,此时未考虑取样的代表性或忽视药品质量管理风险。正确的做法是可以考虑通过过程控制,例如原辅料的质量控制、生产环境控制等实现参数放行而非一味的考虑对终产品做文章。
其次基于风险评估减少终产品检验量或检验项目申报时,需要提交相应的风险评估报告及相应的依据,不能简单的一句经风险评估得出就万事大吉了。
也许是微生物检验人才的缺乏,很多微生物检验员习惯按照化学检验方法开发的方式来对待微生物检验方法开发,忽视微生物需要考虑pH、渗透压、温度、水活度等对微生物生长的影响,导致开发的方法无法通过方法适用性。需要特别注意有些样品经过影响微生物生长或检出的处理时需要做对照实验,而不是仅在最后进行薄膜过滤或者接种平皿前时加试验菌通过实验即认为通过实验。
3)因资料书写不完善导致资料缺少或欠完善导致无法开展实验室工作,此处是最需要避免的。除做好资料审核外,为保证表达清楚、规范,我们需要注意一个完整的检验方法内容包括但不限于以下内容:通用方法、取样量(样品数量)、操作方法(特别是关键参数)、检验项目、限度标准。对于特别的方法还需要注意一些细节,如静置、缓冲液温度等细节。
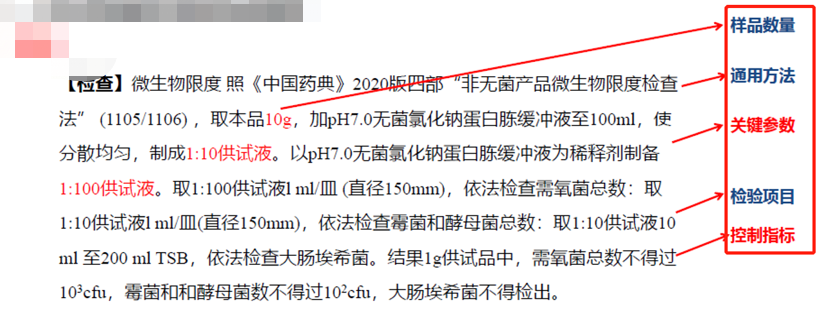
微生物检验方法描述(仅供学习交流用,制药过程中的微生物基础知识 https://www.bopuyun.com/pc/college/207 )
1.2.所有注册产品(实行参数放行或风险管理的出厂放行产品的情况视CDE相关政策)均应有明确的微生物检验标准及对应的方法学资料(或检验SOP)。按《中国药典》制剂通则规定的单一化药成分的部分片剂、胶囊剂、颗粒剂等,微生物限度可以归入“其他”项下,但申报资料扔应有明确的微生物限度标准及方法学资料(或检验SOP)。
思考:
1)对于“实行参数放行或风险管理的出厂放行产品的情况视CDE相关政策”这一段,个人的理解是该部分主要涉及最终灭菌无菌产品参数放行和细胞治疗产品等短效期产品符合这一项要求。
2)他们的共同点都是基于质量风险管理通过过程控制进行放行的经典案例,需要注意一点的是终产品的无菌检查不再是关注重点,但对原辅料、直接与药品接触的包装材料及辅助系统(如气体和润滑剂)微生物状况的评估及必要的控制,以及对灭菌前产品生物负载的控制(包括数量和耐热性)要求更为严格。相应的微生物检验方法验证内容反而更多。
3)关于参数放行申报资料如何书写,可以参考《终端灭菌产品实施参数放行的相关申报资料要求》和《湿热灭菌无菌产品参数放行要求团体标准》。
4)对“单一化药成分的部分片剂”我个人的理解是强调的是主要成分及原料药而不是说整个片剂是单一成分。其次需要强调一下“其他”一般是指通用项目,不是指不重要的项目,重要性和其他逐一列举的项目是一样的,任何一个指标不合格,企业都是不能放行的。
1.3.申请注册无菌制剂及原料药的,其无菌检查的检验数量、检验量应复核所申报药典和中国药典相关规定,且申报标准与对应方法学资料(或检验SOP)应前后一致。控制项目缺失、取样量减少、检验程序变更等又没有详细风险研究资料的或者标准和方法不一致的,将导致无法开展复核实验。
思考:
1)国内与国外无菌检查的培养体系是一样的,只是验证用菌种不同,其次国内需要做阳性对照实验。此处可以吐槽一下做了方法适用性试验还要做阳性对照这一点,会不会与对国内无菌生产企业工艺稳定性存疑有关,并不完全是中国药典委考虑更严谨。
2)对于无菌原料药因为其成本高或产量低等原因,会遇到取样量减少等情况,此时需要注意关注的重点是如何进行评估。评估的前提是基于对产品和产品质量风险的认知,围绕人机料法环进行展开,该部分大家都没有问题,毕竟跟写文献综述比起来也没多难。存在的主要问题是缺少事实依据,也就是历史数据的支持。对历史数据及趋势分析进一步做出前面评估的推论,这样有理有据才能更具有说服力。但问题是申报初期大家都没有足够多的数据,此时国外权威文献或者成果申报的案例等作为借鉴和支持也是一种思路。
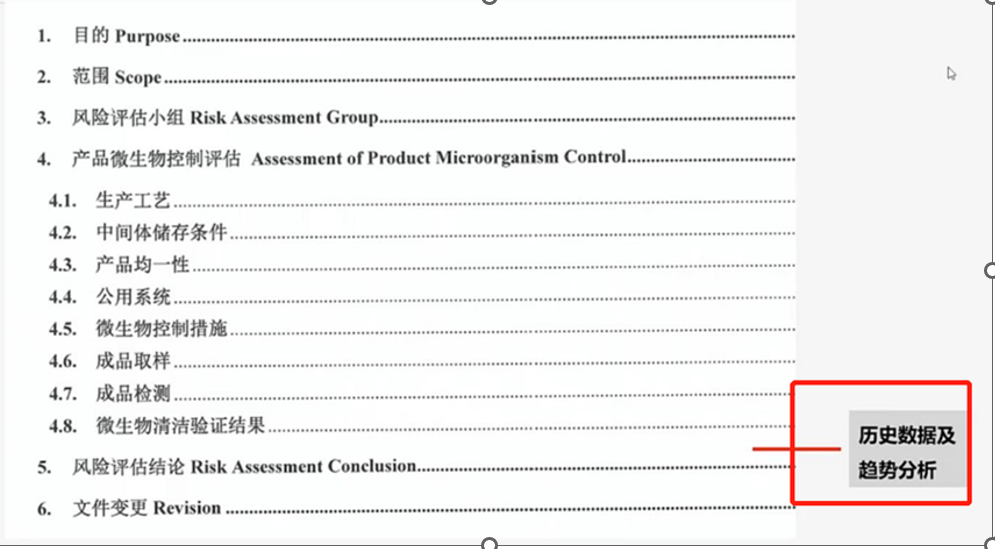
风险评估目录(图片来源于郑老师培训PPT,仅供学习交流用)
1.4.申报方法规定的实验条件无法满足或者存在过度稀释等明显不能满足标准判断需要,且没有任何补救措施的,将无法开展复核实验。
思考:
1)此处就是说了要合规且方法要满足标准判断需要。此处的补救方法需要根据不同的情况进行具体分析,个人没有特别适宜的案例故此处展开,仅举一个例子,某样品抑菌性强,其标准为<100cfu/g,然而1:100的供试品稀释液才可通过过滤消除抑菌性,此时检验1mL就不适宜,至少需要检验10mL。
2)需要特别强调一下“合规”,合规并不是教条的一定要我们照葫芦画瓢,合规的各项要求是基于科学研究和经验积累的结果。关于微生物检验的各项要求,可以看作是良好的检验规范,是为了确保我们的检验结果更真实,避免污染、差错等。
1.5.鉴于当前中国药典和USP/EP等国外药典正在协调过程中,微生物检验标准菌株尚未互认及对等收载,如果申报标准为USP/EP,而方法学资料使用中国药典CMCC菌株的,视为与现实不符合、不真实申报资料,直接退回。反之亦然。

ATCC菌株图片(图片来源于网络仅供参考)
思考:
1)此处告诉我们没有互认前,要申报谁家就用谁家的标准菌株;
2)使用ATCC代替CMCC的需要正视黑曲霉和巴西曲霉不同的现实;
3)某些进口质量标准按中国药典申报,但其申报资料直接欧美翻译的用的ATCC,质量标准和方法验证不一致。
4)据说某些ATCC菌株完成的验证用CMCC菌株复核时会出现复核失败的时候,所以慎之又慎。其次说个题外话就是,中美双报的有多少卖到了国外呢?如果仅是为了某些噱头,长远来说是不适宜的。
2.小结
该部分关于微生物检验方法的附图和风险评估的目录我认为是比较有价值的内容。其他部分内容多为个人分析实践意义不大。接下来一段分享微生物检验申报注册中的问题。
