


网页端用户可点击右方目录直接跳转至制定章节
转发本文到朋友圈,添加下方二维码,免费领取【质量标准编制SOP】


1. 概述
1.1 基本概念
质量标准(Specification):药物的质量研究与质量标准的制订是药物研发的主要内容之一。在药物的研发过程中需对其质量进行系统、深入的研究,制订出科学、合理、可行的质量标准,并不断地修订和完善,以控制药物的质量,保证其在有效期内安全有效。化学药物质量标准建立的规范化过程技术指导原则( 化学药物质量标准建立的规范化过程技术指导原则)
化学药物质量标准建立的规范化过程技术指导原则)
化学药物质量标准建立的规范化过程技术指导原则)The quality standard (i.e., tests, analytical procedures, and acceptance criteria) provided in an approved application to confirm the quality of drug substances, drug products, intermediates, raw materials, reagents, components, in-process materials, container closure systems, and other materials used in the production of a drug substance or drug product. For the purpose of this definition, acceptance criteria means numerical limits, ranges, or other criteria for the tests described.
药品注册标准:国家食品药品监督管理局批准给申请人特定药品的标准,生产该药品的药品生产企业必须执行该注册标准。
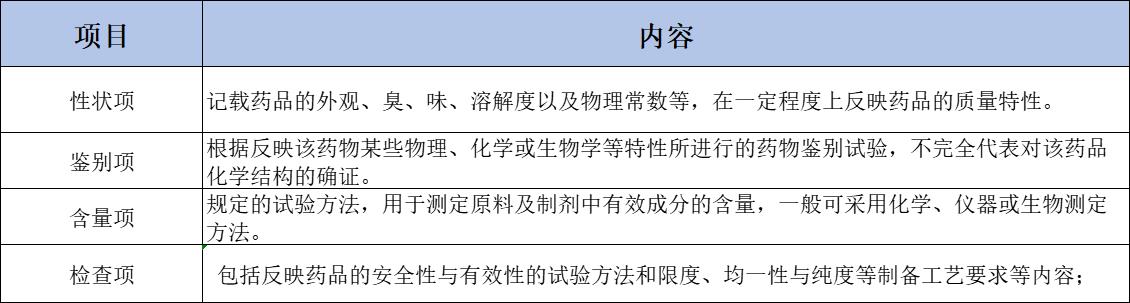
国家药品标准,药典标准:《中华人民共和国药典》简称《中国药典》,依据《中华人民共和国药品管理法》组织制定和颁布实施。《中国药典》一经颁布实施,其同品种的上版标准或其原国家标准即同时停止使用。《中国药典》各品种项下收载的内容为标准正文。 正文系根据药物自身的理化与生物学特性,按照批准的处方来源、生产工艺、贮藏运输条件等所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。具体项目简述如下:
1.2 质量标准分类
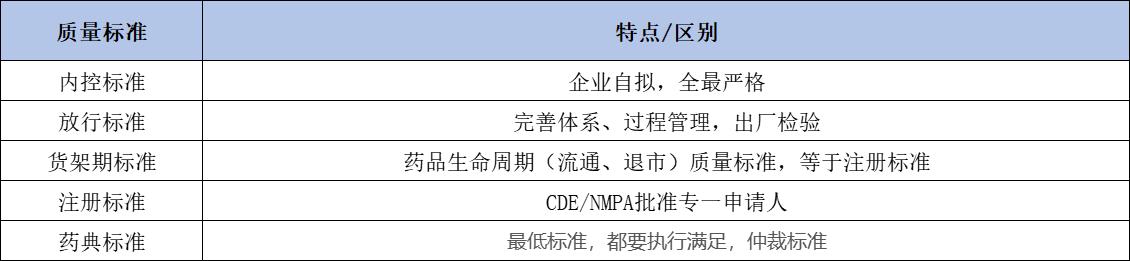
分类:内控标准、放行标准、货架期标准、注册标准、药典标准。
各标准从严程度:内控标准≥放行标准>货架期标准=注册标准>药典标准。
1.3 质量标准制订依据
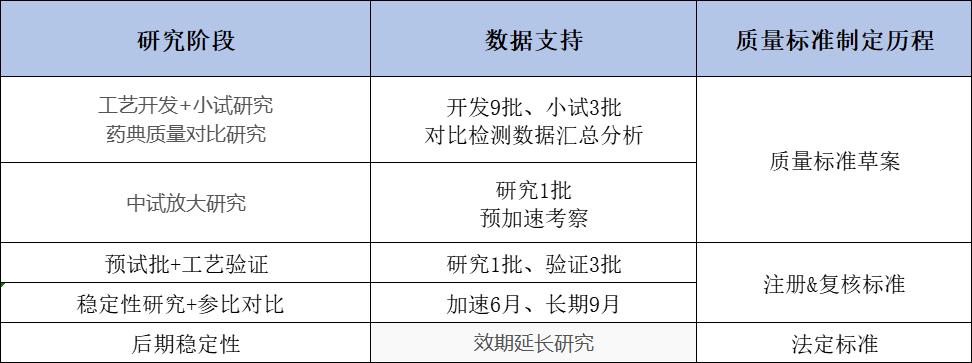
根据工艺开发与研究阶段、工艺验证阶段、稳定性研究阶段质量数据支撑,以及参比制剂质量对比,结合ICH各项研究指南要求,从而制订药品质量标准。
质量标准制订依据数据来源汇总比较:
1.4 质量标准制订历程
草案标准→注册&复核标准→法定标准,分别代表着不同时间段的质量标准形成与使用,层层递进修订与完善。
1.5 各药品类别质量标准制订策略和依据
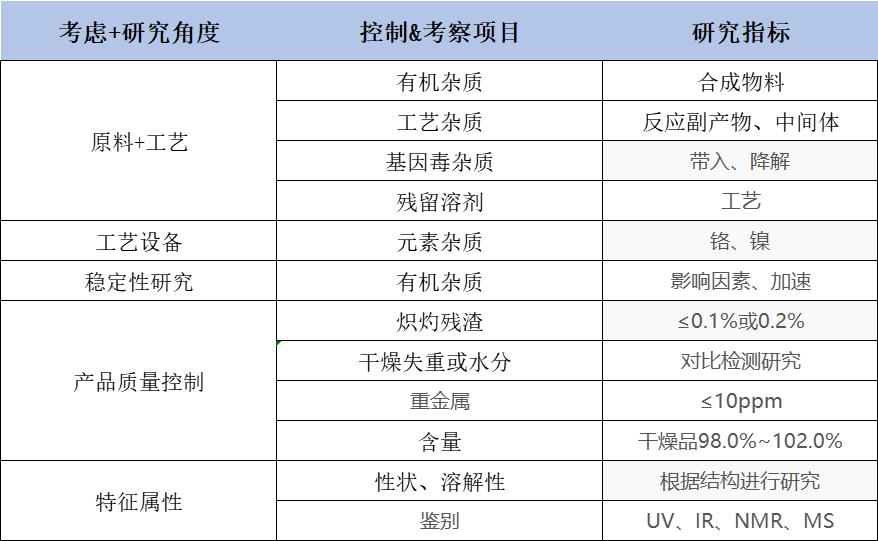
制订依据:主要从原料、工艺、处方、来源、工艺设备、辅助设备、稳定性研究考察数据,综合评估拟定质量标准。其中稳定性研究是最重要的共性指标。
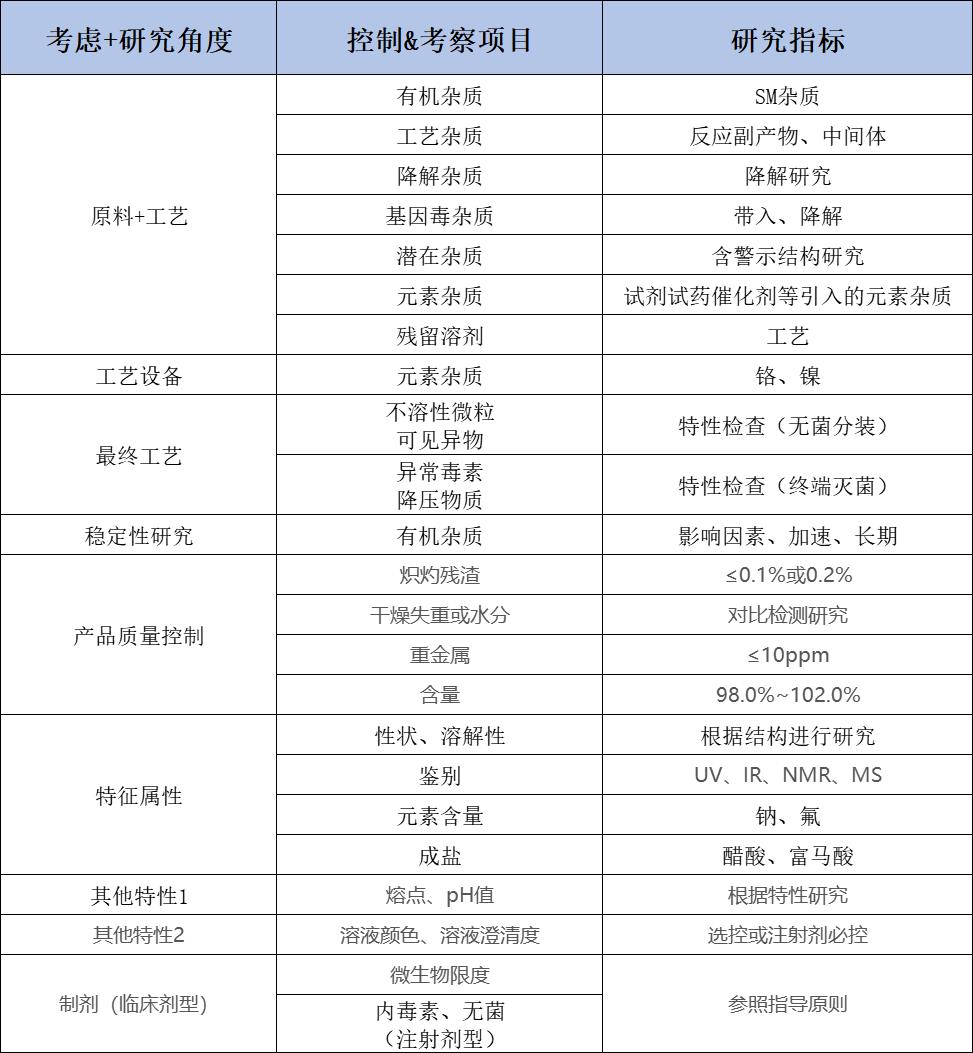
原辅料(包括中药材/提取物)与溶媒制订策略:
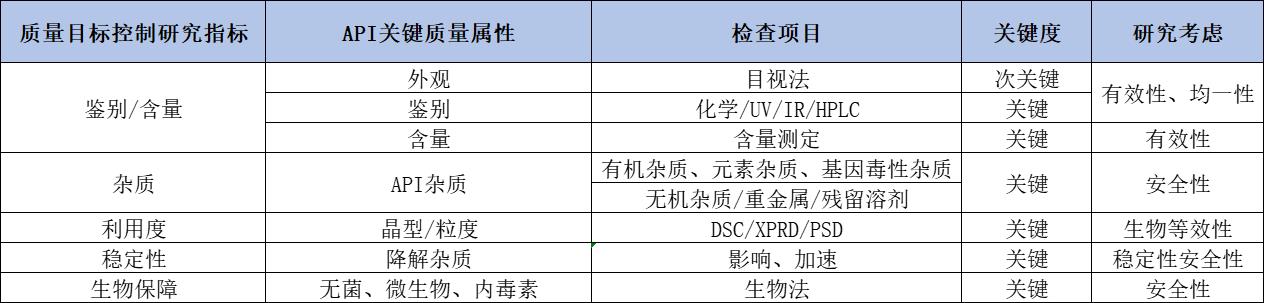
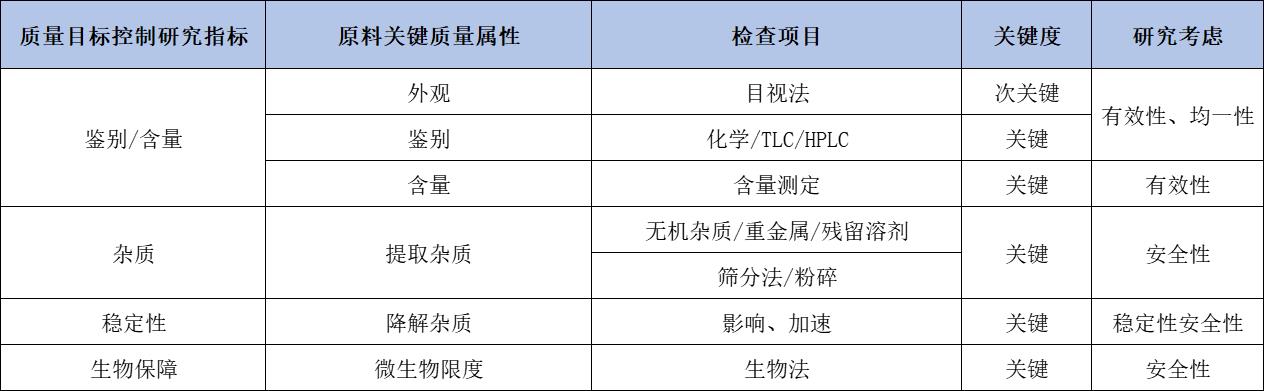
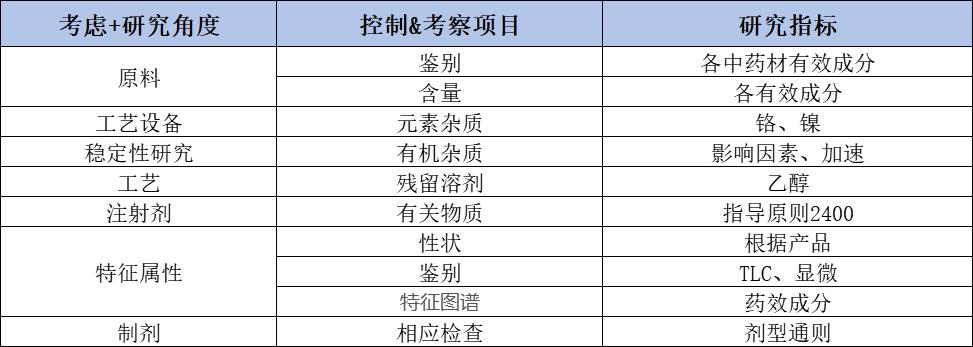
质量标准控制项目制订依据:
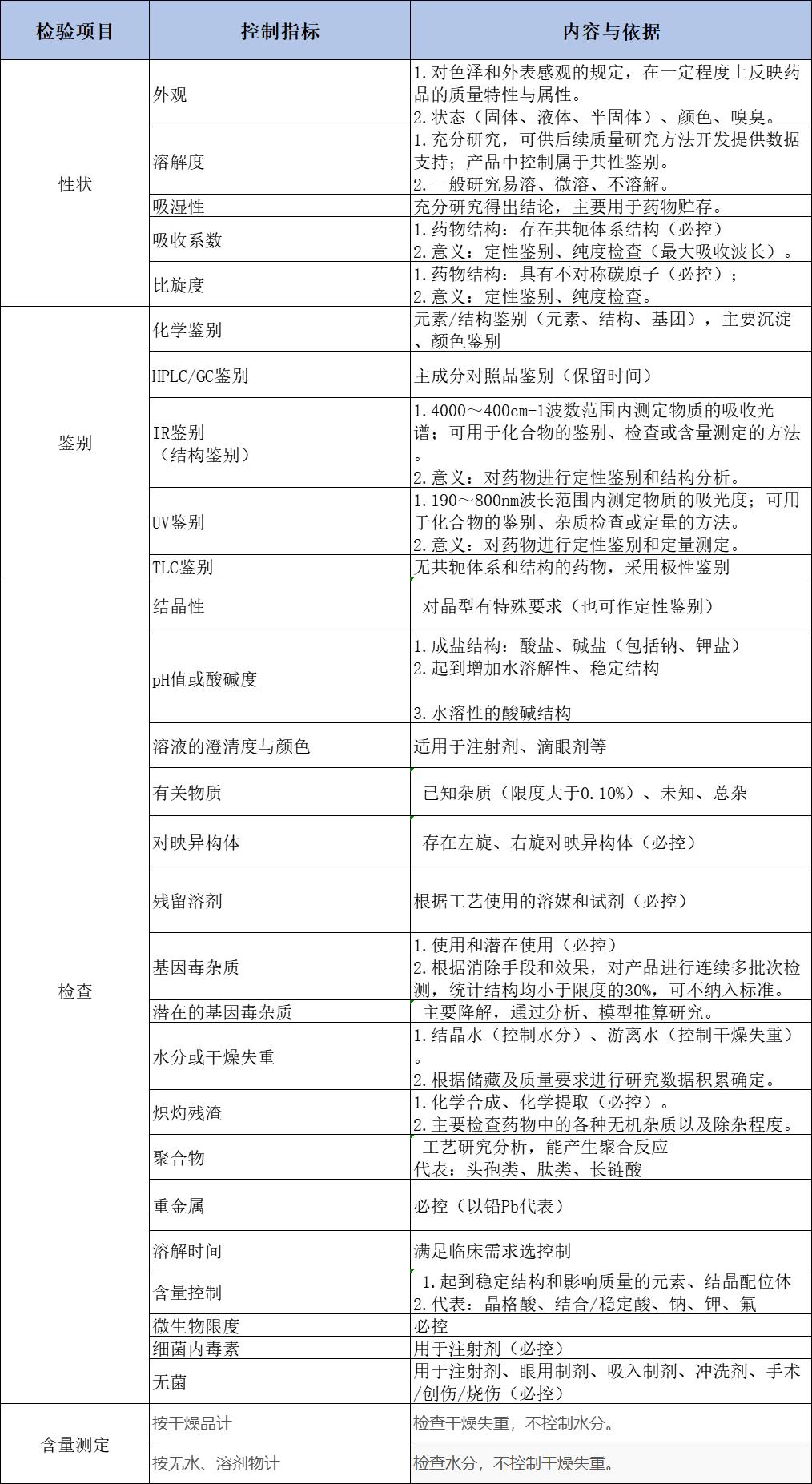
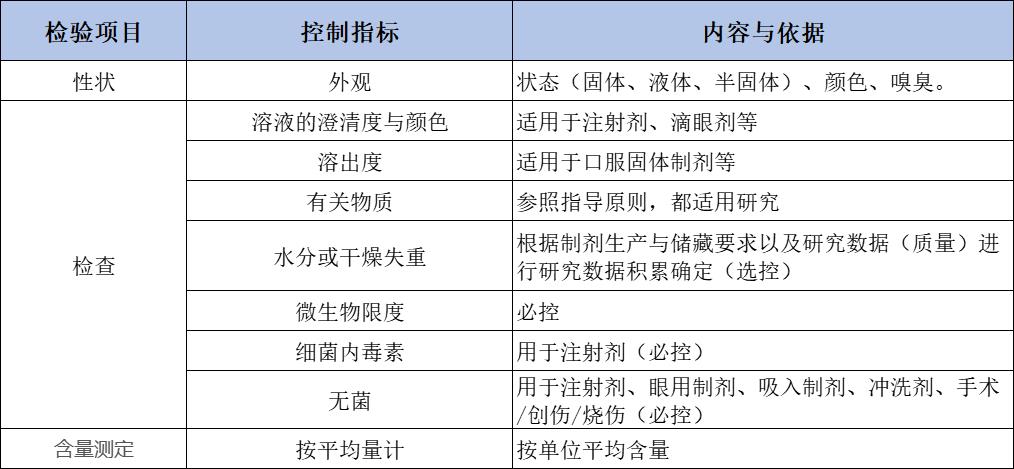
1.6 小结
1.根据并参照上述制订的相关质量标准控制项目,对产品的控制项目(性状、鉴别、检查、含量测定)等方法进行了系统研究,并根据工艺研究及工艺水平、以及产品质量研究,最终根据研究结果制定并建立药品的质量标准。
2.数据支持:实验室放大、转生产批、中试放大、工艺验证、参比制剂对比、稳定性。
2. 法规指南
2.1 国内药监
(药品注册管理办法)
药品注册管理办法)第八条 从事药物研制和药品注册活动,应当遵守有关法律、法规、规章、标准和规范;参照相关技术指导原则,采用其他评价方法和技术的,应当证明其科学性、适用性;应当保证全过程信息真实、准确、完整和可追溯。
药品应当符合国家药品标准和经国家药品监督管理局核准的药品质量标准。经国家药品监督管理局核准的药品质量标准,为药品注册标准。药品注册标准应当符合《中华人民共和国药典》通用技术要求,不得低于《中华人民共和国药典》的规定。申报注册品种的检测项目或者指标不适用《中华人民共和国药典》的,申请人应当提供充分的支持性数据。
药品审评中心等专业技术机构,应当根据科学进展、行业发展实际和药品监督管理工作需要制定技术指导原则和程序,并向社会公布。
第三十九条 综合审评结论通过的,批准药品上市,发给药品注册证书。 综合审评结论不通过的,作出不予批准决定。 药品注册证书载明药品批准文号、持有人、生产企业等信息。非处方药的药品注册证书还应当注明非处方药类别。
经核准的药品生产工艺、质量标准、说明书和标签作为药品注册证书的附件一并发给申请人,必要时还应当附药品上市后研究要求。 上述信息纳入药品品种档案,并根据上市后变更情况及时更新。
药品批准上市后,持有人应当按照国家药品监督管理局核准的生产工艺和质量标准生产药品,并按照药品生产质量管理规范要求进行细化和实施。
第四十四条 化学原料药、辅料及直接接触药品的包装材料和容器关联审评通过的或者单独审评审批通过的,药品审评中心在化学原料药、辅料及直接接触药品的包装材料和容器登记平台更新登记状态标识,向社会公示相关信息。 其中,化学原料药同时发给化学原料药批准通知书及核准后的生产工艺、质量标准和标签,化学原料药批准通知书中载明登记号;不予批准的,发给化学原料药不予批准通知书。
未通过关联审评审批的,化学原料药、辅料及直接接触药品的包装材料和容器产品的登记状态维持不变,相关药品制剂申请不予批准。
第一百六十四条 物料和成品应当有经批准的现行质量标准;必要时,中间产品或待包装产品也应当有质量标准。
第一百六十五条 物料的质量标准一般应当包括:
(一)物料的基本信息:1.企业统一指定的物料名称和内部使用的物料代码;2.质量标准的依据;3.经批准的供应商;4.印刷包装材料的实样或样稿。
(二)取样、检验方法或相关操作规程编号;
(三)定性和定量的限度要求;
(四)贮存条件和注意事项;
(五)有效期或复验期。
第一百六十六条 外购或外销的中间产品和待包装产品应当有质量标准;如果中间产品的检验结果用于成品的质量评价,则应当制定与成品质量标准相对应的中间产品质量标准。
第一百六十七条 成品的质量标准应当包括:
(一)产品名称以及产品代码;
(二)对应的产品处方编号(如有);
(三)产品规格和包装形式;
(四)取样、检验方法或相关操作规程编号;
(五)定性和定量的限度要求;
(六)贮存条件和注意事项;
(七)有效期。
第十二条【标准提高行动计划】 国家实施药品标准提高行动计划,设立专项资金,保障药品标准符合产业高质量发展的需要。
国家药品标准实施后,药品上市许可持有人应当对药品注册标准及时进行评估和修订。
国家鼓励符合规定的药品注册标准转化为国家药品标准。
2.2 FDA
FDA CFR 314.70 Supplements and other changes to an approved application.
(6) The agency may designate a category of changes for the purpose of providing that, in the case of a change in such category, the holder of an approved application may commence distribution of the drug product involved upon receipt by the agency of a supplement for the change. These changes include, but are not limited to: (i) Addition to a specification or changes in the methods or controls to provide increased assurance that the drug substances or drug product will have the characteristics of identity, strength, quality, purity, or potency that it purports or is represented to possess;
FDA CFR 211.165 Testing and release for distribution.
(a) For each batch of drug product, there shall be appropriate laboratory determination of satisfactory conformance to final specifications for the drug product, including the identity and strength of each active ingredient, prior to release. Where sterility and/orpyrogen testing are conducted on specific batches of shortlived radiopharmaceuticals, such batches may be released prior to completion of sterility and/or pyrogen testing, provided such testing is completed as soon as possible.
FDA 21 CFR Sec. 211.110(a) Sampling and testing of in-process materials and drug products
FDA 21 CFR Sec. 211.160 General requirements.
FDA 21 CFR Sec. 211.166 Stability testing.
FDA CFR 211 Sec. 211.192 Production record review.
FDA指南 NDA/ANDA质量标准变更
Specifications (i.e., tests, analytical procedures, and acceptance criteria) are the quality standards provided in an approved application to confirm the quality of drug substances, drug products, intermediates, raw materials, reagents, components, in-process materials, container closure systems, and other materials used in the production of a drug substance or drug product. For the purpose of defining specifications, acceptance criteria are numerical limits, ranges, or other criteria for the tests described.
This guidance list the examples of Major Changes (Prior Approval Supplement), Moderate Changes (Supplement - Changes Being Effected), Minor Changes (Annual Report).
2.3 欧盟
2.4 WHO
2.5 ICH
(ICH Q6A)
ICH Q6A)(ICH Q6B)
ICH Q6B)ICH Q6A Specifications : Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances
ICH Q6B Specifications : Test Procedures and Acceptance Criteria for Biotechnological/Biological Products
3.优质内容
3.1 资源专题
【GMP认证】管理标准-质量标准
 https://www.bopuyun.com/pc/subject/80
https://www.bopuyun.com/pc/subject/80【GMP认证】质量管理-质量标准
https://www.bopuyun.com/pc/subject/1193.2 优质文章
3.3 优质课程
Q6A:新药的质量标准制定
https://www.bopuyun.com/pc/college/lesson/614