


网页端用户可点击右方目录直接跳转至制定章节
转发本文到朋友圈,添加下方二维码,免费领取【药品上市许可持有人制度(MAH)法规汇编全集】

1. 概述
上市许可持有人(Marketing authorisation holder,MAH),药品上市许可持有人是指取得药品注册证书的企业或者药品研制机构等。药品上市许可持有人应当依照本法规定,对药品的非临床研究、临床试验、生产经营、上市后研究、不良反应监测及报告与处理等承担责任。其他从事药品研制、生产、经营、储存、运输、使用等活动的单位和个人依法承担相应责任。
药品上市许可持有人的法定代表人、主要负责人对药品质量全面负责。
药品注册是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出药物临床试验、药品上市许可、再注册等申请以及补充申请,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,决定是否同意其申请的活动。申请人取得药品注册证书后,为药品上市许可持有人。
实施 MAH 制度,给了制药行业更多可操作的空间。但对于原有的制药企业,不应过度反应,如果没有委托事宜,那么就与新制度实施前一样,只需要正常依照法规的更新做好内部的相关工作即可;如果有委托事宜(研发、临床、生产、销售、药物警戒)那么需要做好管理工作,可以委托事项但无法委托责任,最终负责人都是持有人。所选择的受托方要合规、符合相关法规要求;要与受托方建立良好的沟通机制;建立信任关系;签订委托协议、质量协议,明确双方责任与义务;对受托方做好日常监督,对药监部门所要求的报告按时完成,也就是需要建立法规要求的药品质量保证体系,配备专门人员独立负责药品质量管理,履行好 MAH 职责。要做好前述这些工作,是需要依照所有现行法规指南的,但本篇只归纳 MAH 新制度下的主要内容。
2. 中国 MAH 试点相关文件
2015年11月05日,全国人民代表大会常务委员会关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定( 全国人民代表大会常务委员会关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定)(2015年11月4日第十二届全国人民代表大会常务委员会第十七次会议通过)
全国人民代表大会常务委员会关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定)(2015年11月4日第十二届全国人民代表大会常务委员会第十七次会议通过)
全国人民代表大会常务委员会关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定)(2015年11月4日第十二届全国人民代表大会常务委员会第十七次会议通过)第十二届全国人民代表大会常务委员会第十七次会议“授权国务院在北京、天津、河北、上海、江苏、浙江、福建、山东、广东、四川十个省、直辖市开展药品上市许可持有人制度试点,允许药品研发机构和科研人员取得药品批准文号,对药品质量承担相应责任。本决定授权的试点期限为三年。
2016年6月6日,国务院办公厅发布了《关于印发药品上市许可持有人制度试点方案的通知》(《关于印发药品上市许可持有人制度试点方案的通知》)(国办发〔2016〕41号)
《关于印发药品上市许可持有人制度试点方案的通知》)(国办发〔2016〕41号)试点方案包括九个方面,包括试点内容、试点药品范围、持有人及受托生产企业条件等。
2016年06月6日,国家局发布《药品上市许可持有人制度试点方案》(《药品上市许可持有人制度试点方案》)政策解读
《药品上市许可持有人制度试点方案》)政策解读解答了14个热点问题,例如法律依据等。
2016年07月06日,总局关于做好药品上市许可持有人制度试点有关工作的通知(食药监药化管〔2016〕86号)(总局关于做好药品上市许可持有人制度试点有关工作的通知(食药监药化管〔2016〕86号))
总局关于做好药品上市许可持有人制度试点有关工作的通知(食药监药化管〔2016〕86号))提出鼓励符合条件的申请人申报参加试点。
2016年09月29日,国家局发布《药品上市许可持有人制度试点方案》政策解读(二)(国家局发布《药品上市许可持有人制度试点方案》政策解读(二))
国家局发布《药品上市许可持有人制度试点方案》政策解读(二))解答了17个热点问题,例如试点审批主体等。
2017年01月06日,国家局发布《药品上市许可持有人制度试点方案》政策解读(三)
解答了21个热点问题,例如药品研发机构、科研人员作为持有人的,是否可以自行销售所持有品种等。
2017年01月06日,国家局发布《药品上市许可持有人试点品种申报情况》
截至2016年12月25日,共有165个受理号,其中广东和江苏的申请数量居多,临床试验申请的数量远高于上市申请和补充申请。
2017年08月21日 ,总局关于推进药品上市许可持有人制度试点工作有关事项的通知 (食药监药化管〔2017〕68号)
试点内容增加:药品生产企业集团公司集中持有各控股子公司的药品批准文号、药品生产企业因加工场地或车间异地搬迁、整体搬迁或被兼并后整体搬迁成为持有人。
2018年10月27日,全国人民代表大会常务委员会关于延长授权国务院在部分地方开展药品上市许可持有人制度试点期限的决定(全国人民代表大会常务委员会关于延长授权国务院在部分地方开展药品上市许可持有人制度试点期限的决定)(2018年10月26日第十三届全国人民代表大会常务委员会第六次会议通过)
全国人民代表大会常务委员会关于延长授权国务院在部分地方开展药品上市许可持有人制度试点期限的决定)(2018年10月26日第十三届全国人民代表大会常务委员会第六次会议通过)试点工作的三年期限延长一年。
2019年8月26日,中华人民共和国药品管理法(中华人民共和国药品管理法)发布,将于2019年12月1日起施行。
中华人民共和国药品管理法)发布,将于2019年12月1日起施行。国家对药品管理实行药品上市许可持有人制度。药品上市许可持有人依法对药品研制、生产、经营、使用全过程中药品的安全性、有效性和质量可控性负责。
3. 实施文件
3.1 上市前
【MAH资格确认】
【默认自动转为 MAH】
新修订的药品管理法全面实施药品上市许可持有人制度。自2019年12月1日起,凡持有药品注册证书(药品批准文号、进口药品注册证、医药产品注册证)的企业或者药品研制机构为药品上市许可持有人,应当严格履行药品上市许可持有人义务,依法对药品研制、生产、经营、使用全过程中药品的安全性、有效性和质量可控性负责。
【MAH 只能有一个主体】
新修订的《药品管理法》实施后提出的药品注册申请,不再受理两个和两个以上主体共同作为上市注册申请人的上市注册申请,上市注册申请批准后,该注册申请人自然成为上市许可持有人。
对已经受理上市注册申请,涉及两个或两个以上共同申请人的,最晚不得迟于该注册申请技术审评结束前提交注册申请表中所有申请机构签署同意的《上市注册申请人确认书》,确定其中一个主体为该注册申请批准后的上市许可持有人。
【MAH 主体不包括个人】
新药品管理法第三十条 药品上市许可持有人是指取得药品注册证书的企业或者药品研制机构等。
MAH主体不限于药品生产企业,没有生产能力的企业或者药品研制机构也可以申请成为MAH,但与试点方案不同的是,“个人”并未出现。药品上市是一个全生命周期管理的过程,个人无法承担商品交易、纳税等相应的义务,不能对药品研发、生产、流通等环节实施有效管理。
【必备生产许可证】
- (药品注册管理办法)
第五十条 申请药品上市许可时,申请人和生产企业应当已取得相应的药品生产许可证。
MAH 如何获得药品生产许可证?— 根据2020版药品生产监督管理办法,委托他人生产制剂的药品上市许可持有人,应当具备本办法第六条第一款第一项((一)有依法经过资格认定的药学技术人员、工程技术人员及相应的技术工人,法定代表人、企业负责人、生产管理负责人(以下称生产负责人)、质量管理负责人(以下称质量负责人)、质量受权人及其他相关人员符合《药品管理法》《疫苗管理法》规定的条件;)、第三项((三)有能对所生产药品进行质量管理和质量检验的机构、人员;)、第五项((五)有保证药品质量的规章制度,并符合药品生产质量管理规范要求。)规定的条件,并与符合条件的药品生产企业签订委托协议和质量协议,将相关协议和实际生产场地申请资料合并提交至药品上市许可持有人所在地省、自治区、直辖市药品监督管理部门,按照本办法规定申请办理药品生产许可证。
- 需关注各省、自治区、直辖市药品监督管理部门的规定,如天津市关于规范药品生产许可办理工作的通知
- (NMPA 关于实施药品注册管理办法有关事宜的公告 2020年第46号)
新《药品注册管理办法》实施后受理的药品上市许可申请,申请人应当在受理前取得相应的药品生产许可证.
【原料药持有人能否申请成为MAH】
在试点期间,由于当时原料药被视为药品,按照药品进行管理,所以当时是可以成为MAH的。 现在,虽然原料药依然按照药品进行管理,但对原辅包实行关联审评,原料药持有人就不能成为 MAH 了。 提醒行业注意,原料药在我国实行审批制(关联审评审批)和生产许可证管理。
【境外持有人与境内法人】
药品管理法第三十八条 药品上市许可持有人为境外企业的,应当由其指定的在中国境内的企业法人履行药品上市许可持有人义务,与药品上市许可持有人承担连带责任。
3.2 生产质量
- (药品管理法)
- (药品生产监督管理办法)

协议中应明确规定持有人应配备质量受权人,负责产品的最终上市放行。持有人不得将产品的上市放行工作授权给受托方完成。 受托方只能进行产品的出厂放行。 物料的放行可以由持有人授权给受托方的质量管理部门完成,也可在质量协议中进行约定。———— 药品委托生产质量协议指南征求意见稿(药品委托生产质量协议指南征求意见稿)
药品委托生产质量协议指南征求意见稿)【委托生产】
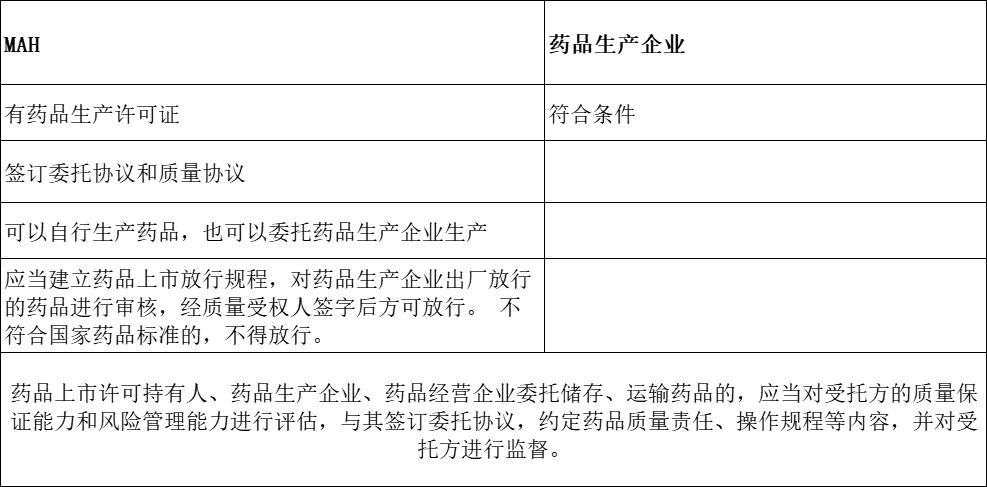
不得委托生产
血液制品、麻醉药品、精神药品、医疗用毒性药品、药品类易制毒化学品不得委托生产;但是,国务院药品监督管理部门另有规定的除外。
经批准或者通过关联审评审批的原料药应当自行生产,不得再行委托他人生产。
【质量授权人】
质量受权人(Qualified Person,Authorized Person)的资质:
应当至少具有药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少五年从事药品生产和质量管理的实践经验,从事过药品生产过程控制和质量检验工作。应当具有必要的专业理论知识,并经过与产品放行有关的培训,方能独立履行其职责。
质量受权人的主要职责:
1.参与企业质量体系建立、内部自检、外部质量审计、验证以及药品不良反应报告、产品召回等质量管理活动;
2.承担产品放行的职责,确保每批已放行产品的生产、检验均符合相关法规、药品注册要求和质量标准;
3.在产品放行前,质量受权人必须按照上述第2项的要求出具产品放行审核记录,并纳入批记录 。中国GMP 第三章 机构与人员(中国GMP 第三章 机构与人员)
中国GMP 第三章 机构与人员)质量受权人(Qualified Person,QP)是中国GMP(2010版)新引入的概念。在欧盟和WHO的GMP中,对质量受权人的相关要求做了明确规定。美国FDA没有相关规定,但其职责被包含在质量管理部门和质量负责人的职责中。下文对欧盟的质量受权人制度做简要介绍。
欧盟质量受权人简介:
在欧盟,质量受权人(Qualified Person,QP)是一种从业资格,由申请人提出申请,监管部门进行审查和注册。
QP不一定是公司雇员,可以自由地与公司签订聘用协议,即在企业生产和质量部门之外,设立第三方的监控体系,这种QP不直接管理企业的质量管理部门,主要对企业的质量管理体系进行审计和监控。
QP主要由各成员国的药监部门或药监部门授权的社会团体进行管理。
3.3 经营管理
【委托经营】
- (药品管理法)
第三十四条
药品上市许可持有人可以自行销售其取得药品注册证书的药品,也可以委托药品经营企业销售。 药品上市许可持有人从事药品零售活动的,应当取得药品经营许可证。
药品上市许可持有人自行销售药品的,应当具备本法第五十二条规定的条件;委托销售的,应当委托符合条件的药品经营企业。药品上市许可持有人和受托经营企业应当签订委托协议,并严格履行协议约定的义务。
药品上市许可持有人销售其取得药品注册证书的药品,应当按照《药品管理法》第三十四条规定开展相关活动。依据《关于药品上市许可持有人试点工作药品生产流通有关事宜的批复(关于药品上市许可持有人试点工作药品生产流通有关事宜的批复)》(国药监函〔2018〕25号)有关规定,在2019年12月1日前,药品上市许可持有人与受托药品生产企业已签订的委托销售合同,在合同期间内受托药品生产企业可继续销售药品,合同到期后不得继续委托药品生产企业销售药品(原则上,药品上市许可持有人委托药品生产企业销售药品不得超过2022年12月31日)。根据《药品管理法》规定,2019年12月1日后,药品上市许可持有人不得与受托药品生产企业签订委托销售合同,签订合同销售的,责令限期整改;逾期不改正的,依据《药品管理法》第一百一十五条处理。
关于药品上市许可持有人试点工作药品生产流通有关事宜的批复)》(国药监函〔2018〕25号)有关规定,在2019年12月1日前,药品上市许可持有人与受托药品生产企业已签订的委托销售合同,在合同期间内受托药品生产企业可继续销售药品,合同到期后不得继续委托药品生产企业销售药品(原则上,药品上市许可持有人委托药品生产企业销售药品不得超过2022年12月31日)。根据《药品管理法》规定,2019年12月1日后,药品上市许可持有人不得与受托药品生产企业签订委托销售合同,签订合同销售的,责令限期整改;逾期不改正的,依据《药品管理法》第一百一十五条处理。- 药品经营许可证管理办法(原国家食品药品监督管理局局令第6号)2004年,继续有效,其中规定与《药品管理法》不一致的,按照《药品管理法》执行。
- 药品流通监督管理办法(原国家食品药品监督管理局局令第26号)2007年,继续有效,其中规定与《药品管理法》不一致的,按照《药品管理法》执行。
- (《药品经营监督管理办法(征求意见稿)》)
- (药品经营质量管理规)范
【药品追溯体系与制度】
- (药品管理法)
第三十六条 药品上市许可持有人、药品生产企业、药品经营企业和医疗机构应当建立并实施药品追溯制度,按照规定提供追溯信息,保证药品可追溯。
第四条 药品上市许可持有人、药品生产企业应当建立并实施药品追溯制度,按照规定赋予药品各级销售包装单元追溯标识,通过信息化手段实施药品追溯,及时准确记录、保存药品追溯数据,并向药品追溯协同服务平台提供追溯信息。
3.4【检查】
3.5 上市后
【药物警戒】
MAH 药物警戒体系建设:
- 招聘具有PV经验的人员;
- 拟定各项制度和附件表格;
- 根据国家局要求申请账户,并按时上报数据;
- 根据省局要求,定期自查并递交报告;
- 持续关注 MAH 涉及的产品 PV 信息
- (《药品不良反应报告和监测管理办法》)
- (国家药品监督管理局关于药品上市许可持有人直接报告不良反应事宜的公告(2018年第66号))
- (个例药品不良反应收集和报告指导原则)
- CDR 药品上市许可持有人药物警戒年度报告撰写指南试行
- (CDR 药物警戒委托协议撰写指导原则(试行))
【变更】
详见【变更】(【变更】)
【变更】)【年度报告】
(药品管理法)
药品管理法)第三十七条 药品上市许可持有人应当建立年度报告制度,每年将药品生产销售、上市后研究、风险管理等情况按照规定向省、自治区、直辖市人民政府药品监督管理部门报告。
【上市许可转让】
第四十条 经国务院药品监督管理部门批准,药品上市许可持有人可以转让药品上市许可。受让方应当具备保障药品安全性、有效性和质量可控性的质量管理、风险防控和责任赔偿等能力,履行药品上市许可持有人义务。
这意味着上市许可产权地位的确立,MAH 拥有许可处置权,可以因为继承、收购、兼并重组、技术转让等申请转让许可。因此,药企可以与各医药大学、科研院所建立合作关系,受让 MAH 转让的药品上市许可;或者接受委托生产经营新研发的药品,降低药品研发成本,加快科技成果转化。
(药品注册管理办法)
药品注册管理办法)持有人转让药品上市许可,持有人应当以补充申请方式申报,经批准后实施
4.优质内容
4.1 资源专题
MAH制度解读与学习文章汇编
 https://www.bopuyun.com/pc/subject/183
https://www.bopuyun.com/pc/subject/183MAH(药品上市许可持有人制度)制度相关资料
https://www.bopuyun.com/pc/subject/1824.2 优质课程
MAH委托生产协议注意事项
https://www.bopuyun.com/pc/college/85新药品管理法要求下的MAH质量体系建立和完善
https://www.bopuyun.com/pc/college/50MAH制度下物料及产品的放行管理和要求
https://www.bopuyun.com/pc/college/60