


1.对照品
提供研究过程中使用的所有对照品(包括主成分对照品和杂质对照品)的相关资料。药典对照品:应提供购买的证明资料、样品照片、标签复印件,并明确来源、批号、纯度等信息。对于其他来源的外购对照品或者自制对照品,应提供相应的来源证明材料(或制备工艺、检验报告)、结构确证资料、质量标准、含量和纯度标定过程等。
2.包装材料
(1)以表格的方式列出包装材料类型、来源及相关证明文件:
表XX:包材类型、来源及相关证明文件

注:关于包材类型,需写明结构材料、规格等。
如铝塑泡罩包装,组成为:PVC/铝、PVC/PE/PVDC/铝、PVC/PVDC/铝;
复合膜袋包装,组成为:聚酯/铝/聚乙烯复合膜袋、聚酯/低密度聚乙烯复合膜袋。
表中包装材料(1)、(2)是指不同的包装材料。提供包材的检验报告(可来自包材生产商或供应商)。
(2)说明原研药或参比制剂使用的内包材,本品内包材是否与其一致,如不一致,应提供所用包材的支持性研究与文献依据。
(3)如有处方工艺改变,详细提供本品与内包材相容性研究的资料,包括相容性试验的内容、每一项研究内容采用的试验设计、考察指标、检测方法及方法学验证、样品制备方法、试验结果及对结果的分析等。相容性研究可以参考国内外相关指导原则进行。如未进行包材相容性研究,应说明理由,并提供充分的文献依据。
3.稳定性
有处方工艺改变的品种,提交申报资料时至少需提供三批中试规模[注]及以上批次样品的6个月的加速试验和12个月的长期试验数据,样品的有效期和贮存条件将根据长期稳定性研究的情况最终确定。未改变处方工艺的品种,仅提供三批样品长期稳定性结果。
[注]中试规模的生产设备的操作原理与材质、原辅材料的质控要求、处方工艺及流程等均应与商业化生产一致,且批量至少为商业化生产规模的十分之一。
稳定性总结
总结所进行的稳定性研究的样品情况、考察条件、考察指标和考察结果,对于修改处方工艺的品种提出贮存条件和有效期。
(1)试验样品
稳定性试验用样品应具有代表性,且至少应为中试规模及以上批次的样品。
表XX:试验样品的相关信息
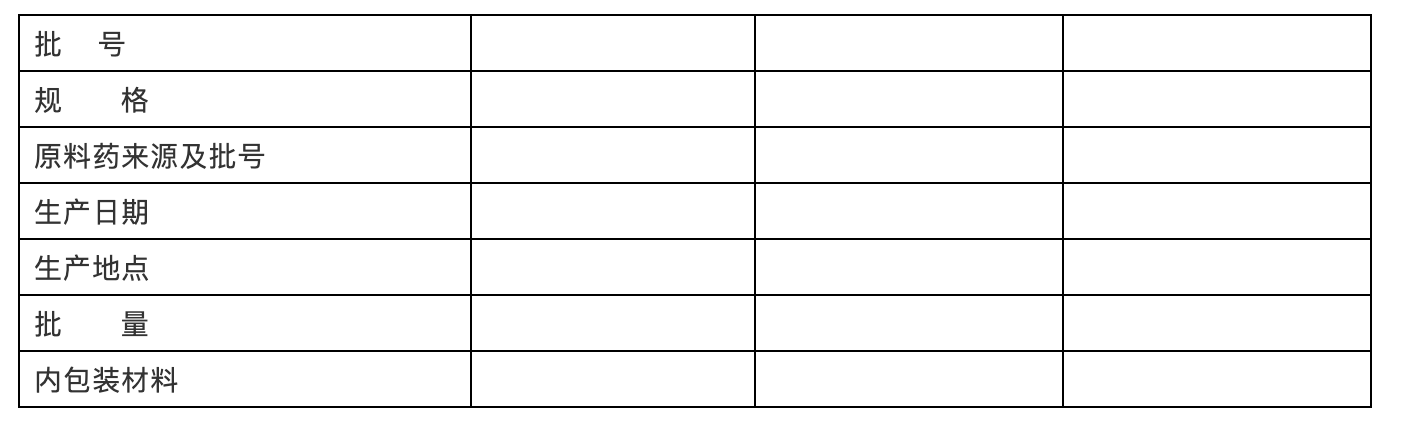
(2)研究内容
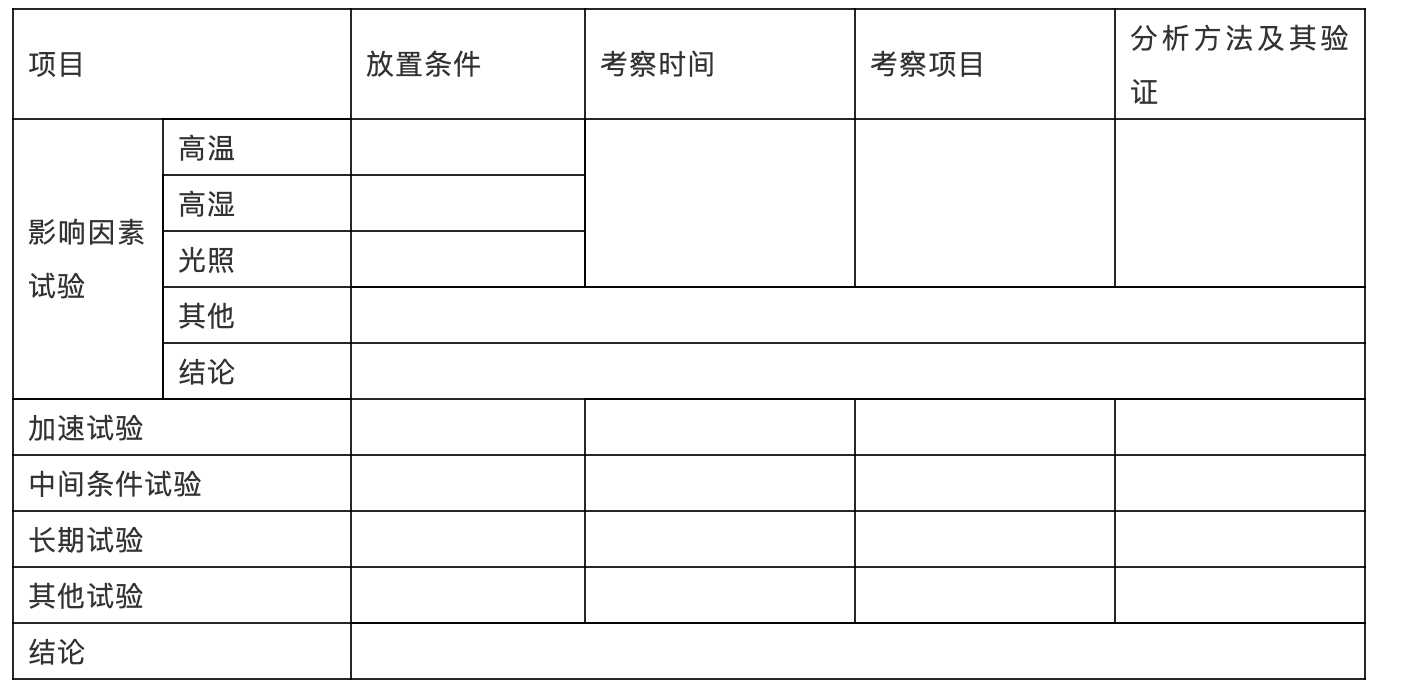
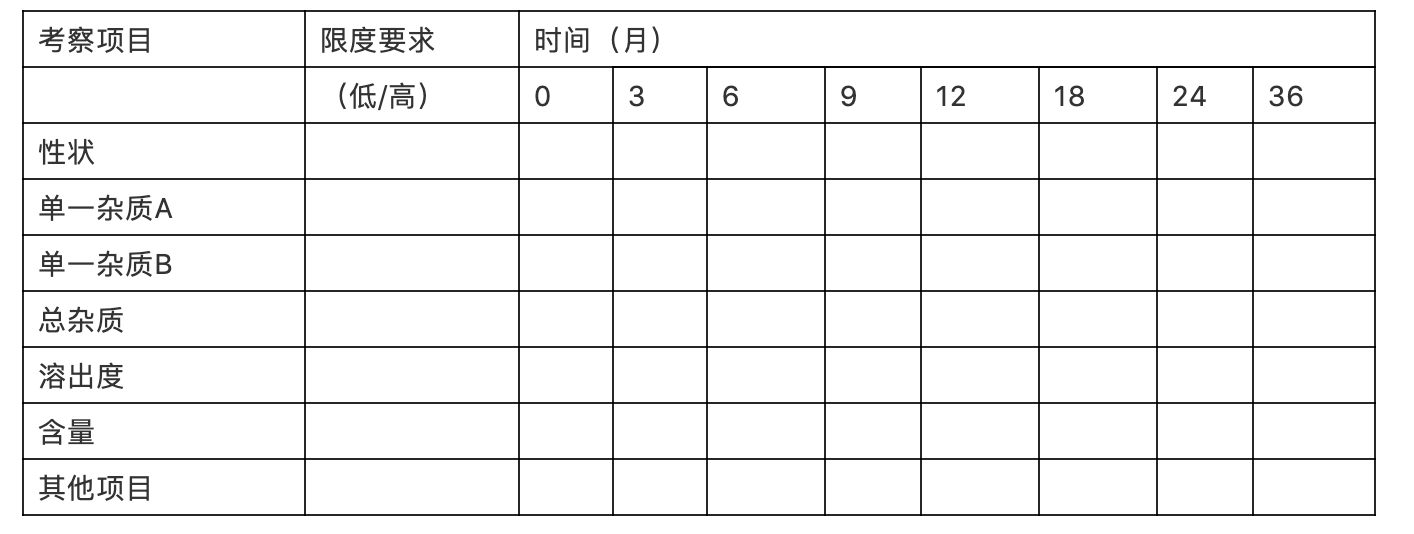
表XX:常规稳定性考察结果
填表说明:
1)影响因素试验中,尚需将样品对光照、高湿、高温之外的酸、碱、氧化和金属离子等因素的敏感程度进行概述,可根据分析方法研究中获得的相关信息,从产品稳定性角度,在影响因素试验的“其他”项下简述;影响因素试验的“结论”项中需概述样品对光照、温度、湿度等哪些因素比较敏感,哪些因素较为稳定,作为评价贮藏条件合理性的依据之一。
2)稳定性研究内容包括影响因素试验、加速试验和长期试验,根据加速试验的结果,必要时应当增加中间条件试验。建议长期试验同时采用30±2℃/65±5%RH的条件进行,如长期试验采用30±2℃/65±5%RH的条件,则可不再进行中间条件试验。
“其他试验”是指根据样品具体特点而进行的相关稳定性研究,如注射剂进行的容器密封性试验等。
3)“分析方法及其验证”项需说明采用的方法是否为已验证并列入质量标准的方法。如所用方法和质量标准中所列方法不同,或质量标准中未包括该项目,应在上表中明确方法验证资料在申报资料中的位置,并在申报资料中说明原因,提供详细的分析方法及其验证资料,以证明该分析方法的可行性。
表XX:使用中产品稳定性研究结果
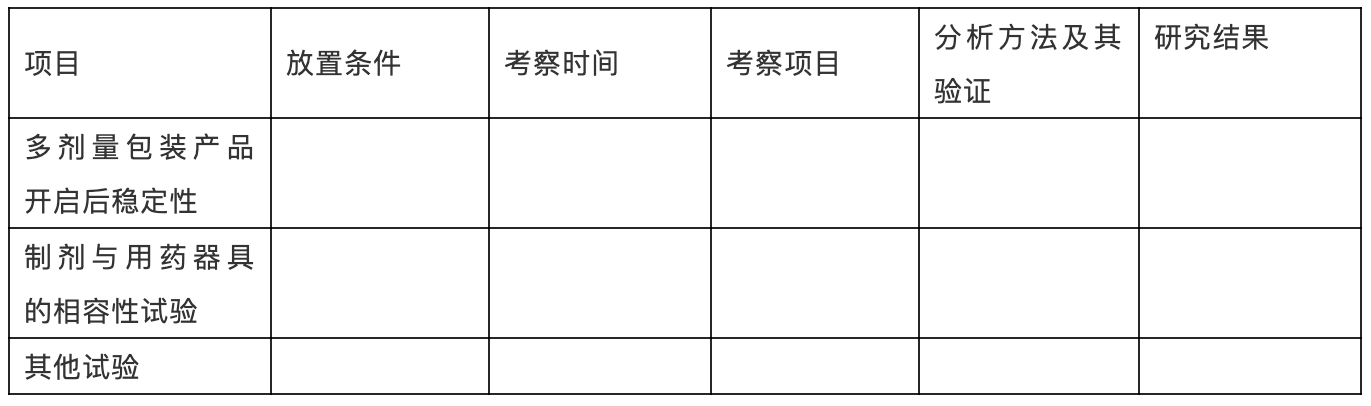
(3)研究结论
总结所进行的稳定性研究的样品情况、考察条件、考察指标和考察结果,对变化趋势进行分析,提出贮存条件和有效期,并与原研药或参比制剂及药典收载的同品种的要求进行比较,被评价品种的稳定性不得更差。示例如下:

后续稳定性承诺和稳定性方案
本部分内容适用于处方、工艺有改变的品种。应承诺对批准后生产的前三批产品进行长期留样稳定性考察,并对每年生产的至少一批产品进行长期留样稳定性考察,如有异常情况应及时通知管理当局。提供后续稳定性研究方案。
稳定性数据
详细提供稳定性考察的相关资料(包括样品的全检报告复印件、加速与长期留样时样品放置的具体地点及恒温柜编号、各时间点质量考察用样品的具体数量等),并以表格形式提供稳定性研究的具体结果,稳定性研究中的相关图谱可作为附件。色谱数据和图谱提交要求参见附件(色谱数据和图谱提交要求)。
(1)影响因素试验(一批样品)
表XX:影响因素试验结果
批号: 批量: 规格:

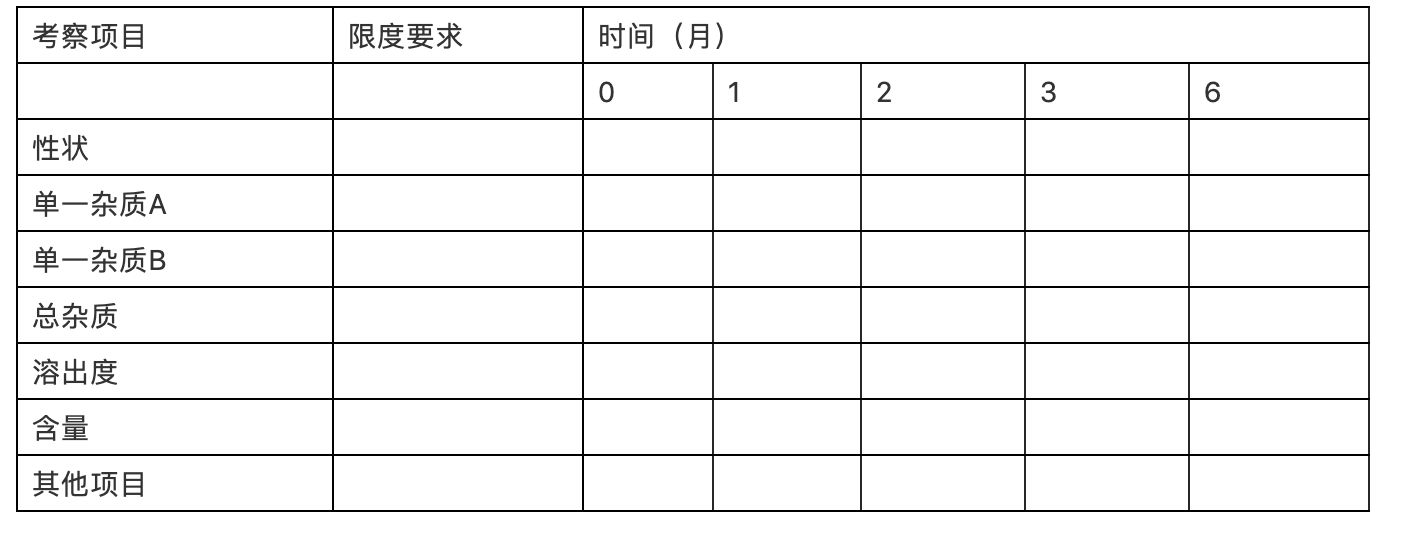
(2)加速试验(三批样品)
表xx:加速试验结果
(批号: 批量:规格: 包装: 考察条件:)
(3)长期试验(三批样品)
表xx:长期试验结果
(批号: 批量:规格: 包装: 考察条件:)
详细提供对长期留样稳定性考察中出现的质量标准中未控制的超过鉴定限度的杂质所做的进一步研究(包括该杂质的结构确证研究、安全性研究等)的资料与图谱等,并提供该杂质限度确定的充分依据(如与原研或参比制剂近效期产品的杂质谱对比研究资料、该杂质的安全性研究资料及现行版国内外药典收载的同品种对该特定杂质的限度要求等)。
4.体外评价-参比制剂
参比制剂的选择
按照《普通口服固体制剂参比制剂选择和确定指导原则》的要求,选择和确定参比制剂。
详述参比制剂选择、备案和审核确认情况,并根据查阅文献或专利信息资料,提供参比制剂处方组成以及生产工艺概述(尽可能了解其特殊的、关键的工艺技术)、辅料与内包材情况,以及对参比制剂的考察等。
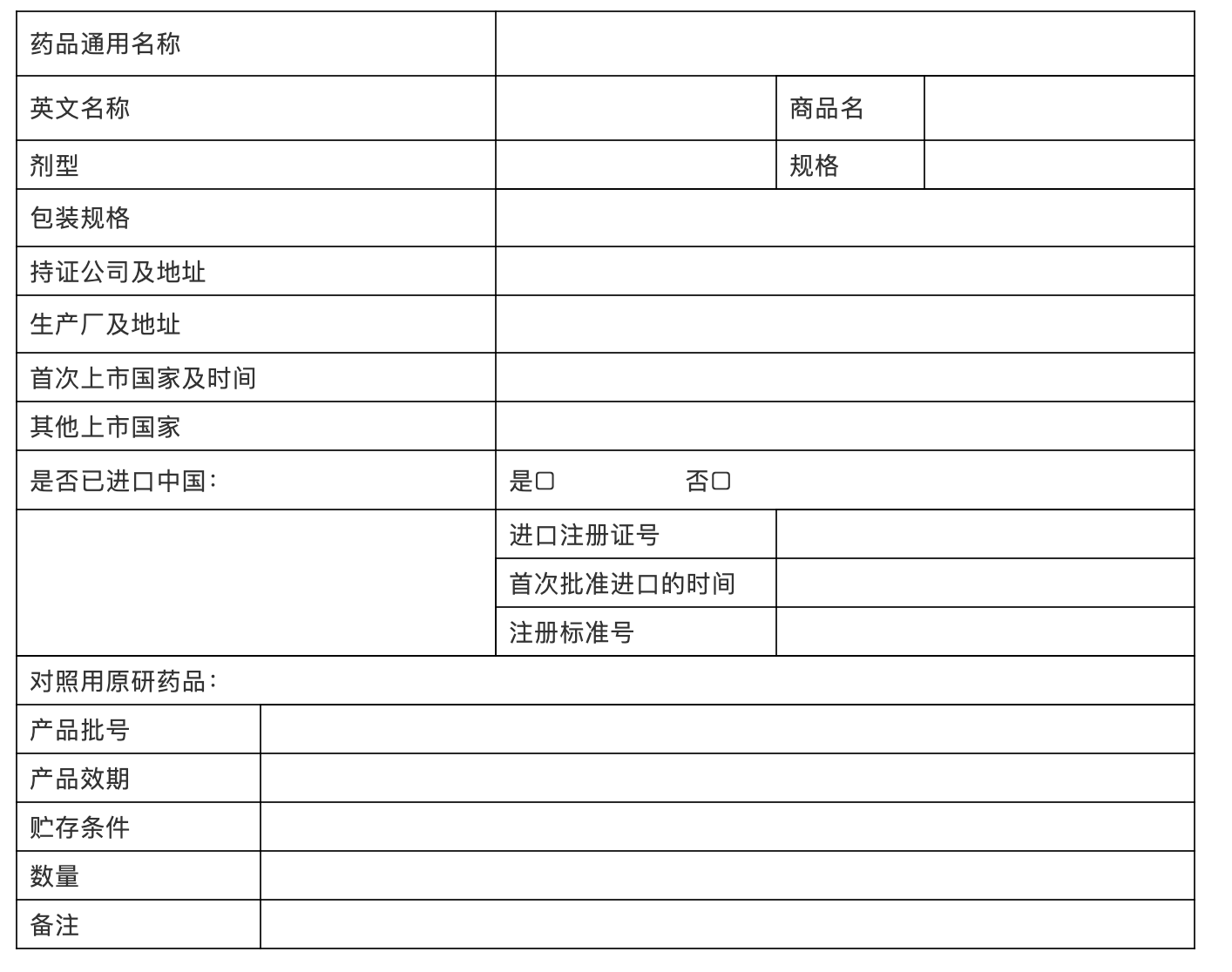
基本信息
提供参比制剂的生产企业名称、批号、规格等,以附件形式提供获得途径以及相关证明性文件(包括官方网站的批准信息、药品说明书、购买发票、赠送证书以及参比制剂实样、标签图片等等)。
表xx: 参比制剂信息表
质量考察
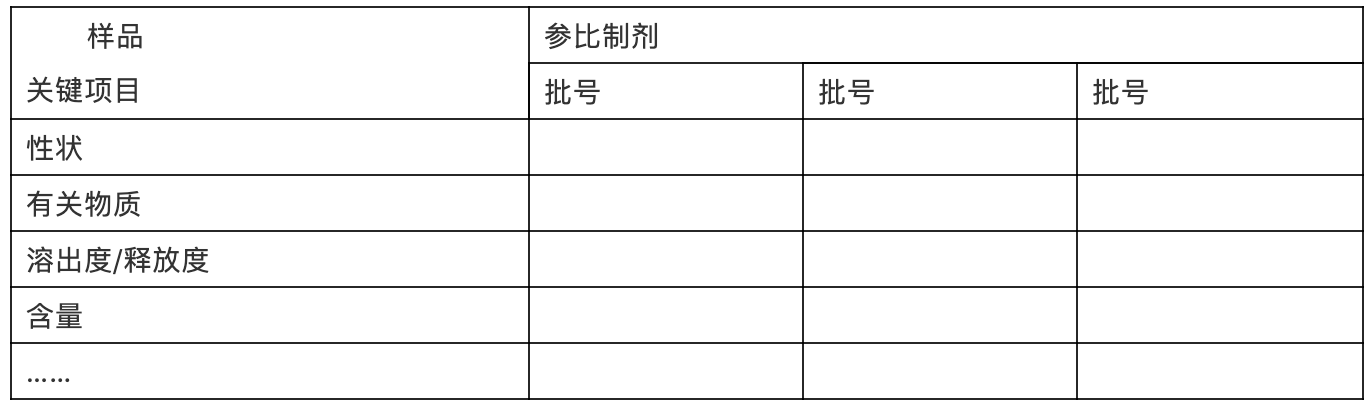
每个规格原则上应提供3批(至少1批)参比制剂的考察数据,考察与一致性评价紧密相关的关键质量属性,例如性状、晶型(原料)、水分、溶出度/释放度、含量、有关物质等(检验报告可列为附件)。以表格形式整理,示例如下:
表xx:参比制剂质量考察研究结果
溶出曲线考察
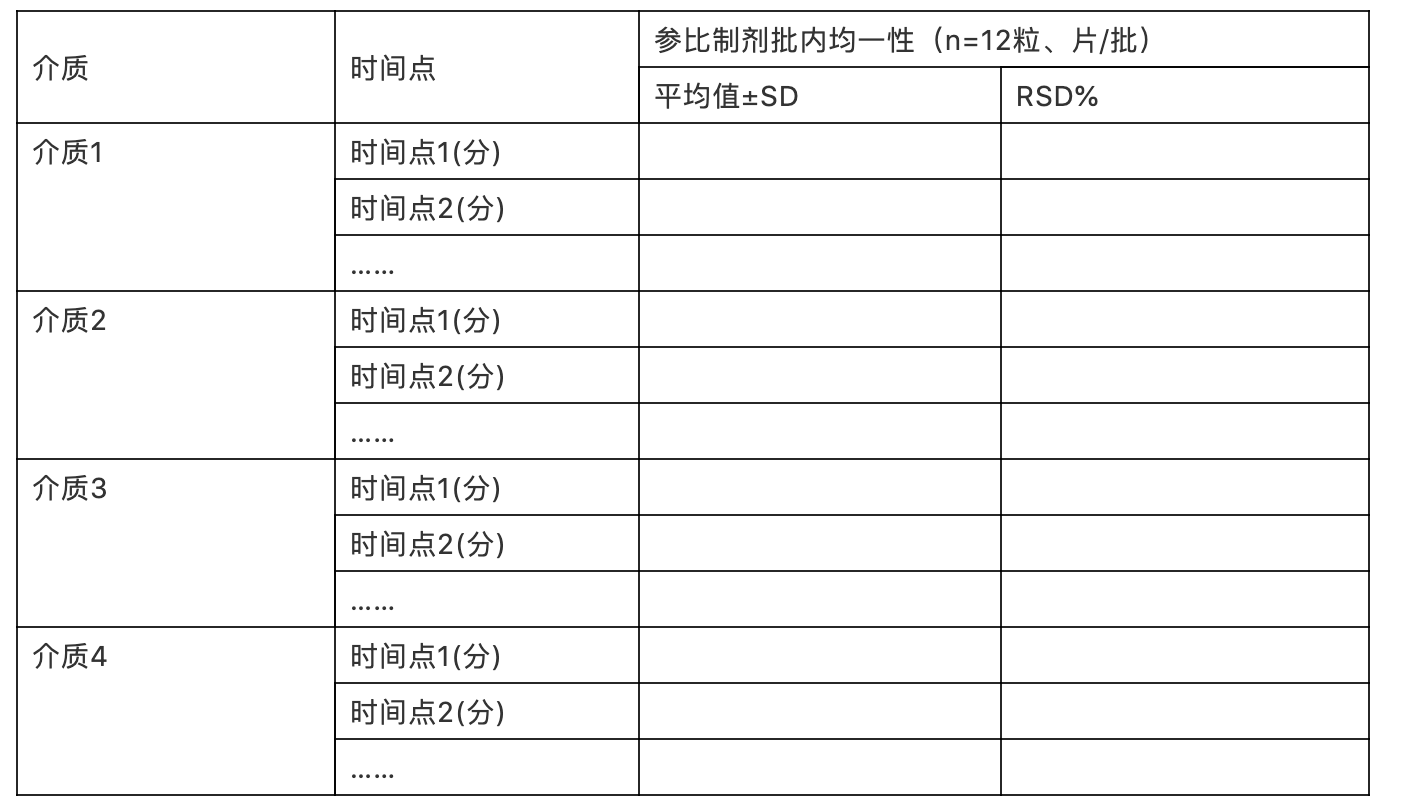
每个规格的参比制剂原则上应提供3批样品的溶出曲线考察数据,以考察其溶出行为的批内和批间均一性,必要时可以平均值、标准偏差与仿制制剂进行相关统计分析。以表格形式整理,示例如下:
表xx:参比制剂批内均一性考察结果(批号:)

溶出曲线稳定性考察
本部分内容适用于理化性质不稳定品种。
对有文献报道或者研究资料表明有光照、高湿、高温、氧化等条件下不稳定的品种,建议考察参比制剂溶出曲线稳定性,为实验室复核结果的重复性提供支持。
取一批参比制剂样品,以企业收到参比制剂的时间作为零时间点(t0),置加速试验条件、长期试验条件或参比制剂说明书中的储存条件下,于零时间点后1月、2月、3月、6月依次取样,并将每个取样时间点的溶出曲线结果与零时间点(t0)比较,结果应相近,如有差异,应在方法误差可接受的范围内。如变化较大,应根据具体情况调整试验方法,并将相应的试验方法和注意事项与复核单位沟通,保证复核结果的一致。申报资料要求提交至少3个月加速试验和3个月长期试验的溶出曲线数据以及结论,以表格形式整理。
建议申报单位在长期试验条件下同时将参比制剂以及仿制制剂留样保存,直至药检机构复核试验结束或至有效期止。
5.质量一致性评价
质量标准比较
应提供充分的试验资料与文献资料,证明仿制制剂的质量与已上市原研产品或参比制剂的质量是一致的,仿制制剂的货架期标准是合理可行的。
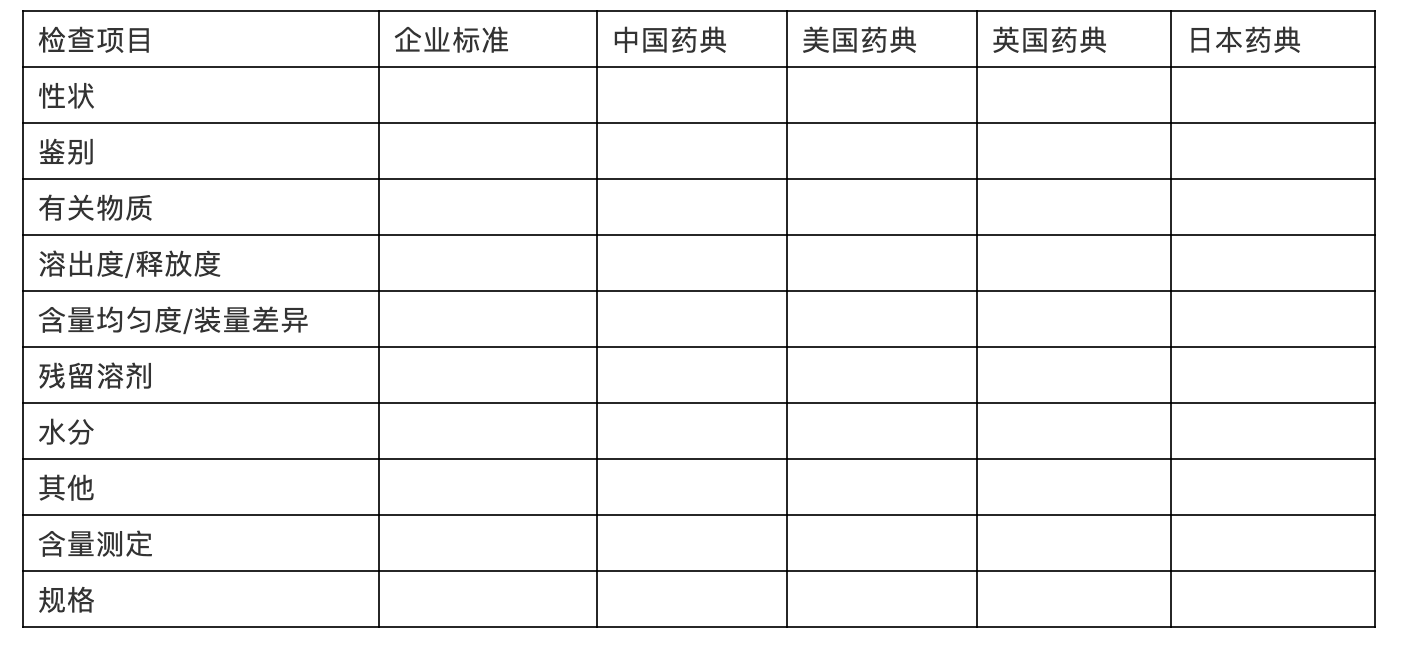
表xx:现行版国内外药典质量标准收载情况。
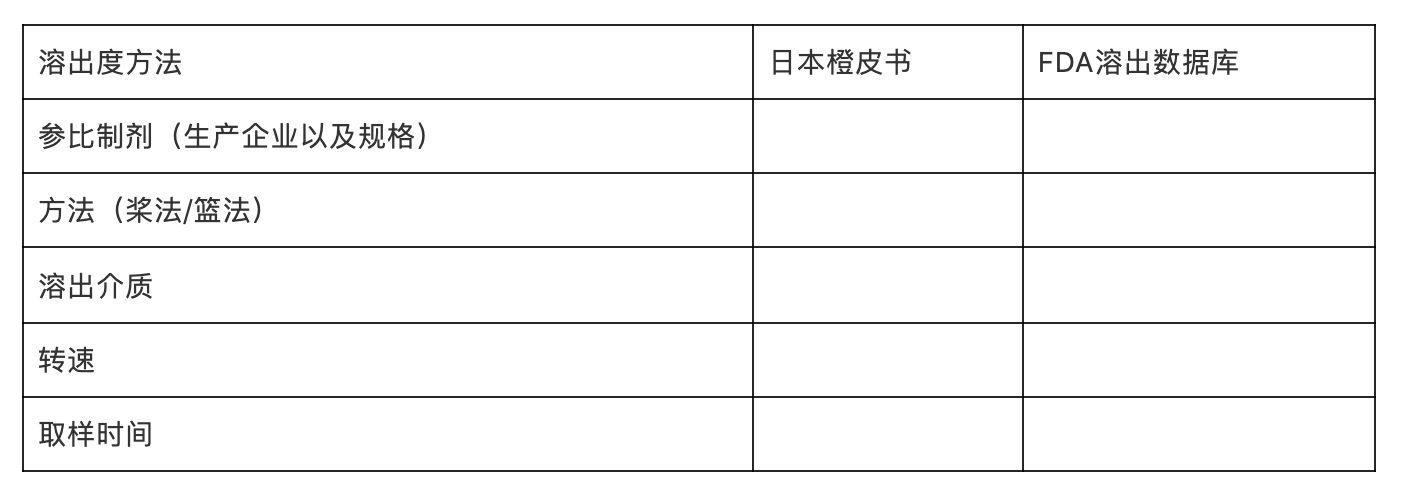
关键质量属性研究(影响一致性评价的关键参数)
通过开展药品质量研究,找出被评价品种与参比制剂相比影响质量和疗效一致性评价的关键参数,例如杂质分析、晶型等。
参比制剂与被评价制剂的检验结果
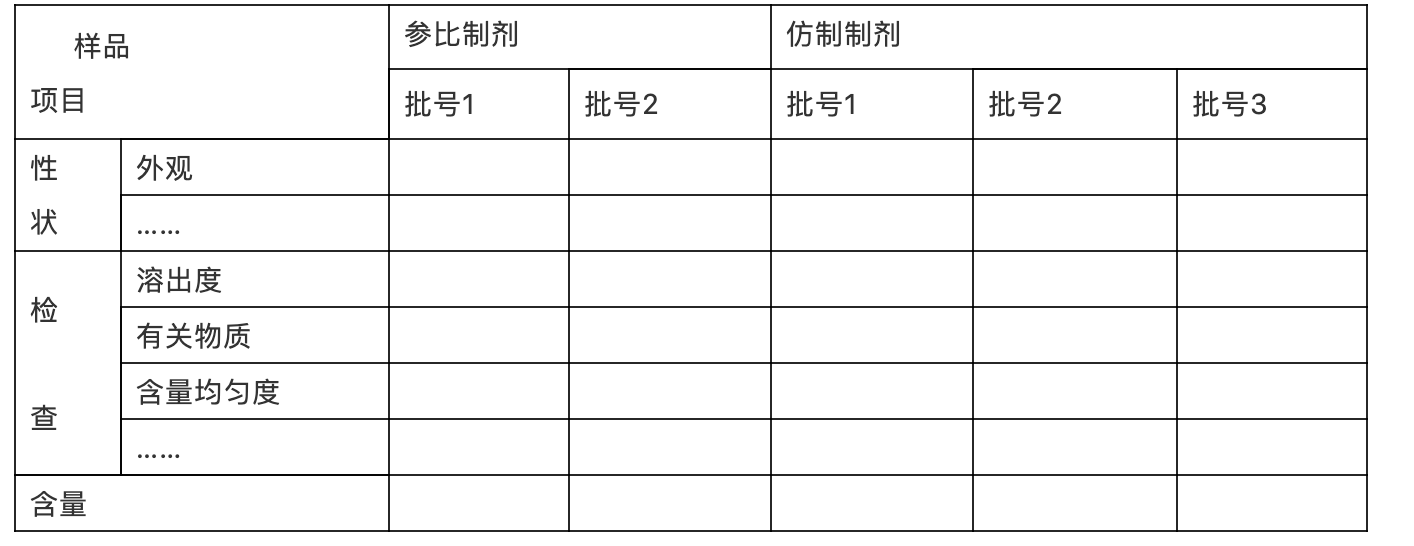
提供被评价制剂和参比制剂进行质量对比研究的资料及结果,以充分证明仿制制剂的质量与参比制剂质量的一致性。以表格形式整理,示例如下:
表xx:仿制制剂与参比制剂整体质量对比研究结果
详细提供与参比制剂杂质比较研究的结果,可参照参比制剂的方法限度考察,以降解产物的量比较制剂稳定性的差异。以表格形式整理,示例如下:
表xx:仿制制剂与参比制剂杂质对比研究结果
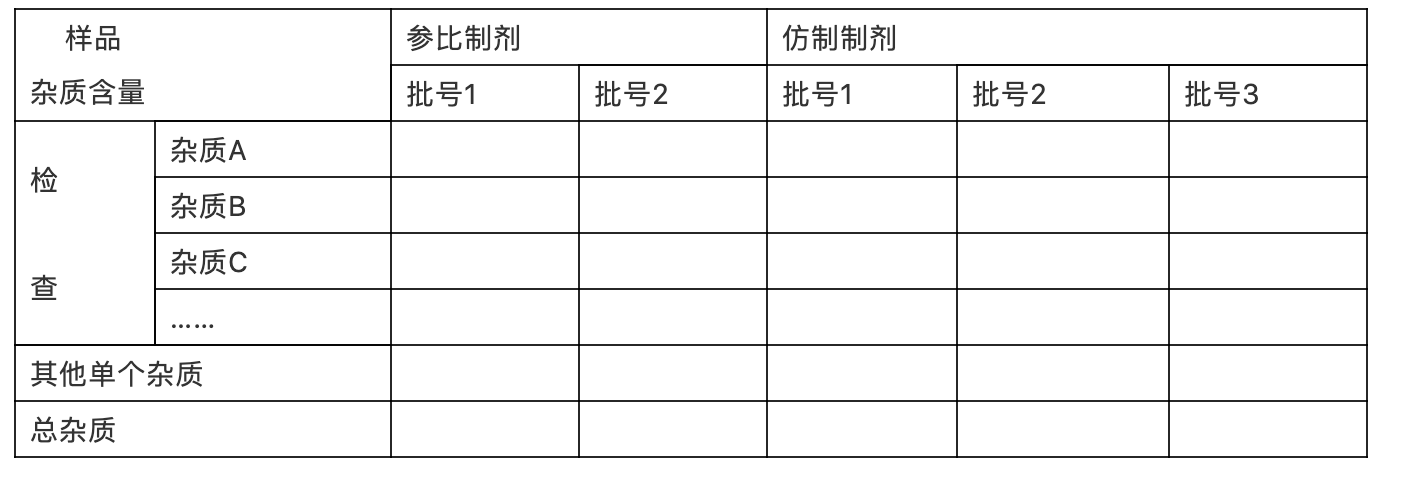
明确是否有超出鉴定限度的新杂质,并按照国内外相关指导原则的要求对这些杂质进行必要的定性研究,新杂质含量不高于参比制剂质量标准或者各国药典标准中其他单个未知杂质限度。若各杂质含量高于参比制剂质量标准或者各国药典标准中其他单个未知杂质限度,应给予充分的合理解释。
References:
[1]《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求》2010修订版
