《中国药典》中规定了无菌注射液、注射用无菌制剂和滴眼剂的可见异物标准,但对于片剂等非无菌制剂以及原辅料中的异物,并无太多参考。片剂通则中仅有一句“片剂外观应完整光洁,色泽均匀。”
企业在制定内控标准,时常无所适从。因大多数原辅料中的可见异物,并未得到充分的研究,安全性没有足够的数据保证,标准的限度更是无从评估。
The International Pharmaceutical Excipients Council(国际辅料协会,IPEC)2015年发布的Technically Unavoidable Particle Profile Guide(技术上不可避免的颗粒指南)ACTIVE PHARMACEUTICAL INGREDIENTS COMMITTEE(欧盟原料药委员会,APIC)2015年6月发布的Guidance on Handling of Insoluble Matter and Foreign Particles in APIs(原料药中不溶物和异物处理指南)
首先,TUPP指南虽然是IPEC针对于辅料制定的,但其结构同样适用于API(辅料在可见异物方面可能较API对制剂的风险更高,影响更大)。因此原辅料都可以参考TUPP指南进行评估和管理。
TUPP指南的宗旨是鼓励采用基于风险的方法评估辅料中的可见异物颗粒。指南的主要内容可以概括为以下几点:
1.要求辅料生产企业基于不同辅料的复杂程度,制定并完善“技术上无法避免的可见异物档案”,并对可能存在的异物类型进行风险评估。
2.辅料生产企业应确定过程中的可见异物的来源,并采用控制策略和技术,并定期评估措施的有效性。
3.当发现超出TUPP档案范围的非典型可见异物时,需要进行全面调查。但是,如果异物与制造设备,设施,垫片,润滑剂等的构造材料一致,则应将其修订入TUPP档案中。
4.鼓励辅料制造商与制剂生产者(用户)之间的沟通,以减少时间,金钱和资源消耗,并确保进行充分的调查。
5.当制剂生产者遇到可见异物时,应联系辅料制造商识别该异物及其来源。辅料制造商提供的TUPP档案可以帮助制剂生产者进行风险评估。制剂生产者应结合制剂的特性和不同剂型需要评估TUPP对制剂的影响。
APIC的<Guidance on Handling of Insoluble Matter and Foreign Particles in APIs>指南,主要针对于API的不溶和外来异物,该指南和IPEC指南有交叉也有互补性,同时虽然APIC指南针对于原料药,但是其指南结构上也适用于辅料。
具体方法就是参考清洁残留允许10 ppm残留限度,假设允许可见异物颗粒有10ppm的残留,根据纤维、硅胶、金属等材质的密度,以及不同粒子的形状,可以计算出异物颗粒的重量,推算出在一定重量的样品内允许存在的可见异物颗粒的数量。
100g的某样品中,10ppm相当于1mg已知金属的密度为8g/cm3,换算为8000kg/m3
如果样品中存在直径为0.5mm的金属颗粒物,则其半径为0.00025m
m=4/3πr3=4/3×π×(0.00025m)3×8000kg/m3=5.24E-07kg=0.524mg
最终结果:100g的该样品中,最多允许存在2粒0.5mm的金属颗粒异物
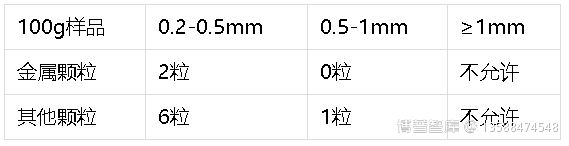
为了达到简化计算的目的,把异物粒子分为2种不同类别:金属(密度:8 g/cm3)和其他粒子(密度:2.5 g/cm3),使用一个球体模型分别使用10ppm的基本限度计算,最终得到的简化限度表:
假设某片剂内控片面异物标准为3%,则每1000片中允许有30粒异物。
假设该片剂片重为0.2g,则0.1g×1000=200g,也就是200g原辅料样品里允许有30粒异物颗粒,100g样品粒允许有15粒异物颗粒。
2.考虑到最严条件,假设原辅料里的异物颗粒都被压在了片剂的外层,或者都在其他剂型的目检范围内。
3.同一片剂中不同比例的原辅料采用一样的限度,实际上可以根据占比再进一步进行折算。如上例中,若该片剂中某一辅料的占比为20%,若假设其他物料中均不会引入异物颗粒,则该物料的异物颗粒限度可以为100g里允许有15/20%=75粒异物颗粒。
可见异物在工艺中不可避免,可见异物的多少反映了企业设备的可靠性、操作的规范性和GMP的符合性。
包括FDA在内的官方监管机构,也并没有把发现可见异物作为缺陷条款,而调查不充分不完整才是真正的问题所在。
所以文中两个指南的理念应该被更多人接受,原辅料生产企业更应该对异物进行充分的重视,并以现有技术能力对异物展开研究并对其进行风险评估,最终为制剂产品的质量评估起到相应的作用。