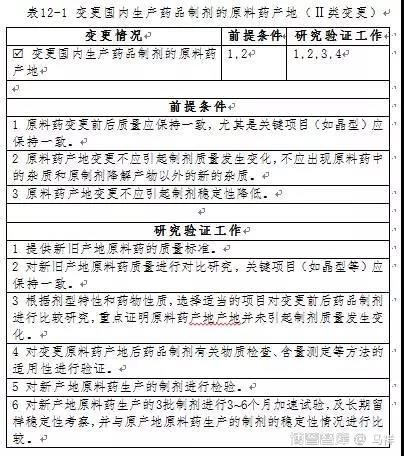
1、具体变更情况及前提条件
这种变更包括改变原料药的生产地点,或增加原料药的生产地点,但原料药的关键理化性质(如晶型等)需保持一致;变更原料药产地后制剂质量不得发生变化,包括药物溶出/释放行为,或与体内吸收和疗效有关的重要理化性质和指标应保持一致,不应出现原料药中的杂质和原制剂降解产物以外的新的杂质;变更原料药产地后制剂稳定性不得较原制剂降低。
2、研究验证工作
根据变更的具体情况,需进行相应的研究验证工作(见表12-1)。
研究工作需证明变更前后原料药主要指标,尤其是原料药的晶型等与变更前保持一致。
同时,需根据剂型特性和药物性质,选择适当的项目对变更前后药品制剂进行比较研究,重点证明原料药产地产地并未引起制剂质量发生变化,药物溶出/释放行为,或与体内吸收和疗效有关的重要理化性质和指标应保持一致,具体工作可参照本指导原则第四章“变更药品处方中已有药用要求的辅料”中Ⅱ类变更研究验证工作相关内容进行。
不同产地原料药合成工艺不同的,引入的工艺杂质也不同,而药品制剂有关物质检查方法及含量测定方法都是基于原产地原料药确定的,所以这些方法是否仍适用新产地的原料药,尚需进行必要的方法学验证,根据验证结果考虑对方法进行修订完善。如标准中检查方法发生了变更,需按本指导原则相关章节进行研究。原则上,原料药产地变更不应出现原料药中的杂质和原制剂降解产物以外的新的杂质。